Yang Bo, Schaefer Anthony J, Small Brooke L, Leseberg Julie A, Bischof Steven M, Webster-Gardiner Michael S, Ess Daniel H
Department of Chemistry and Biochemistry, Brigham Young University Provo Utah 84602 USA
Research & Technology, Chevron Phillips Chemical 1862 Kingwood Drive Kingwood Texas 77339 USA
Chem Sci. 2024 Oct 22;15(44):18355-63. doi: 10.1039/d4sc03433c.
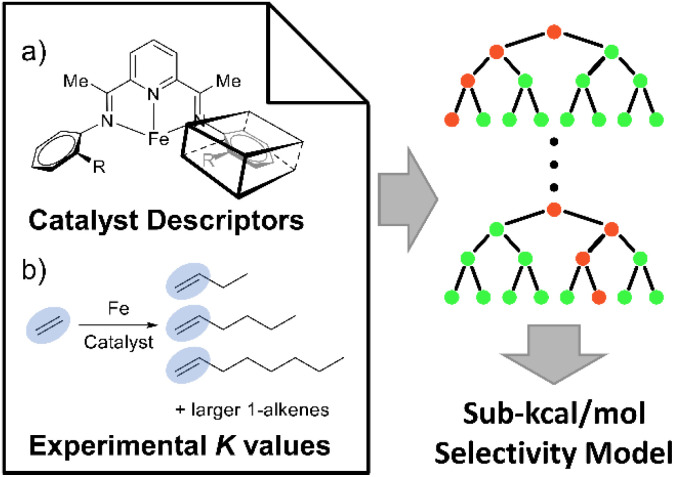
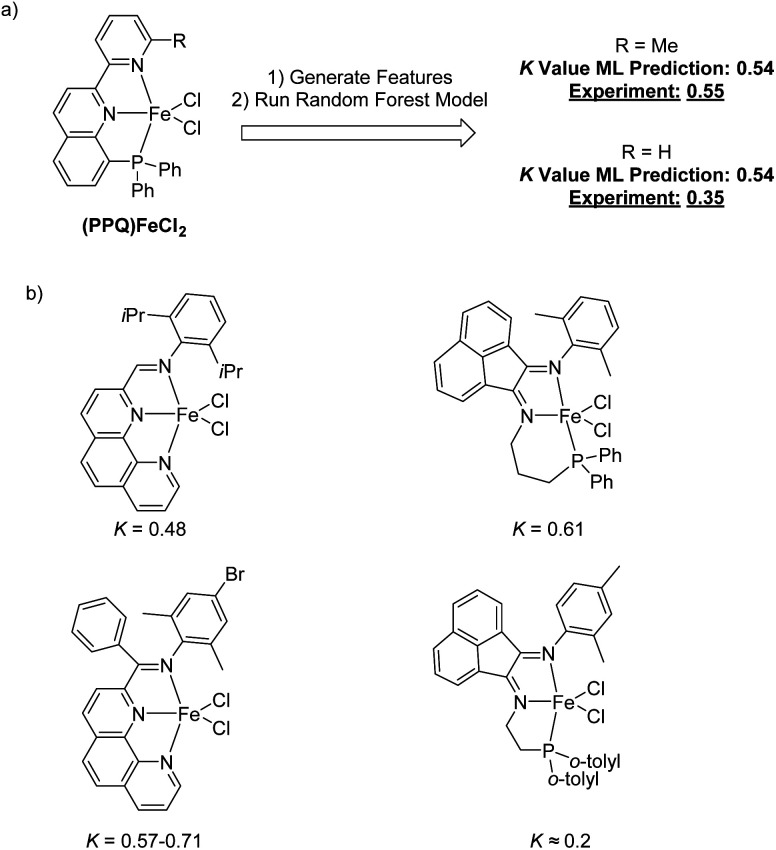
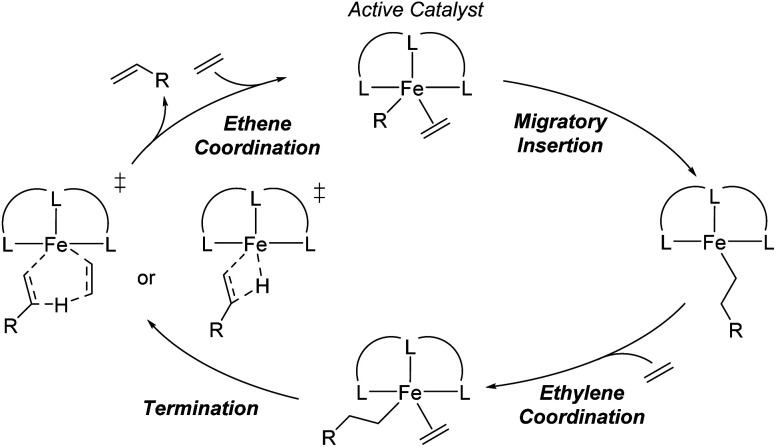
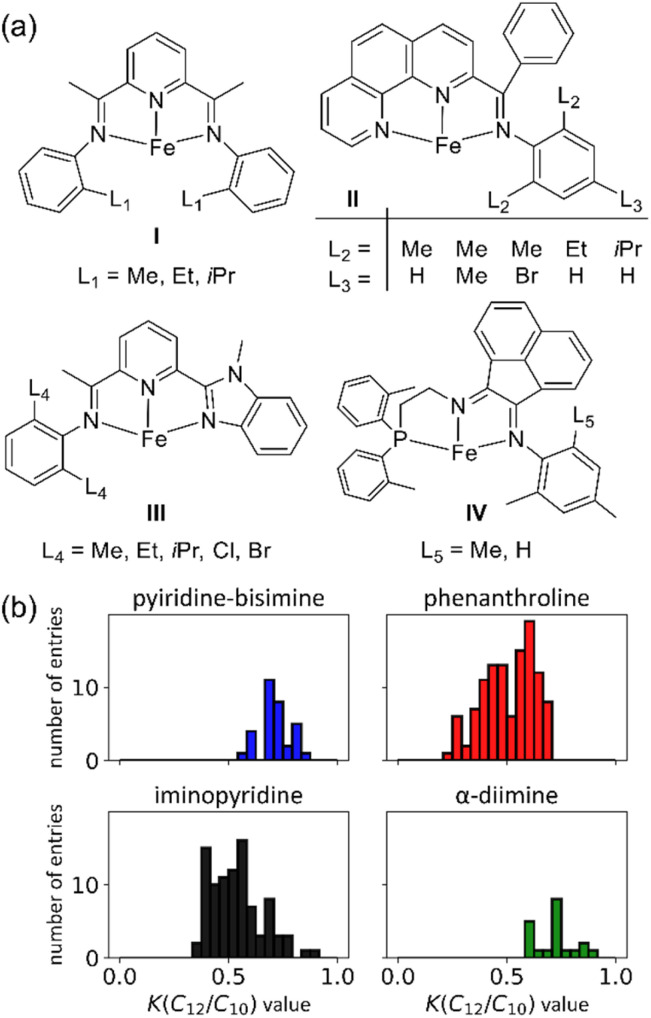
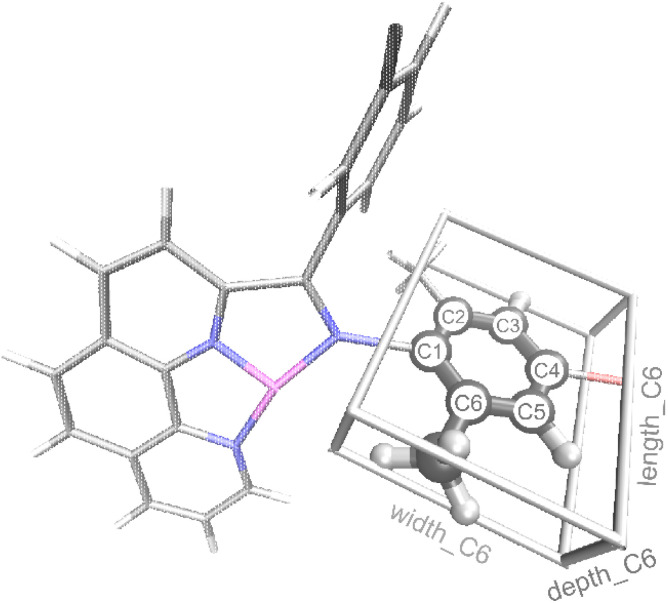
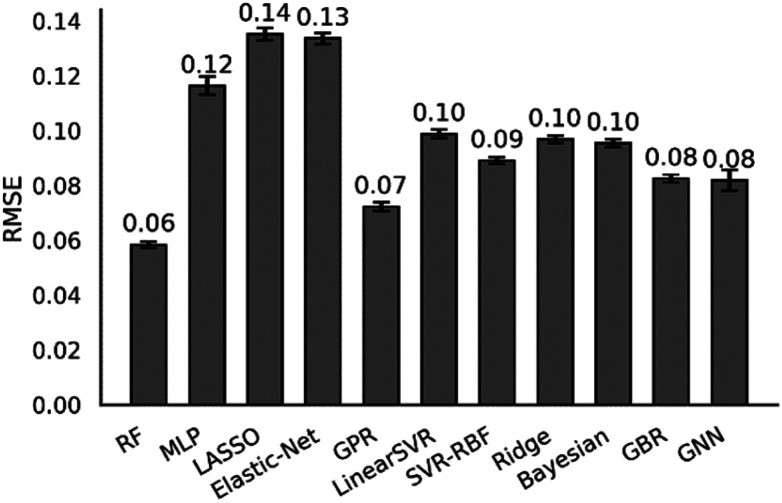
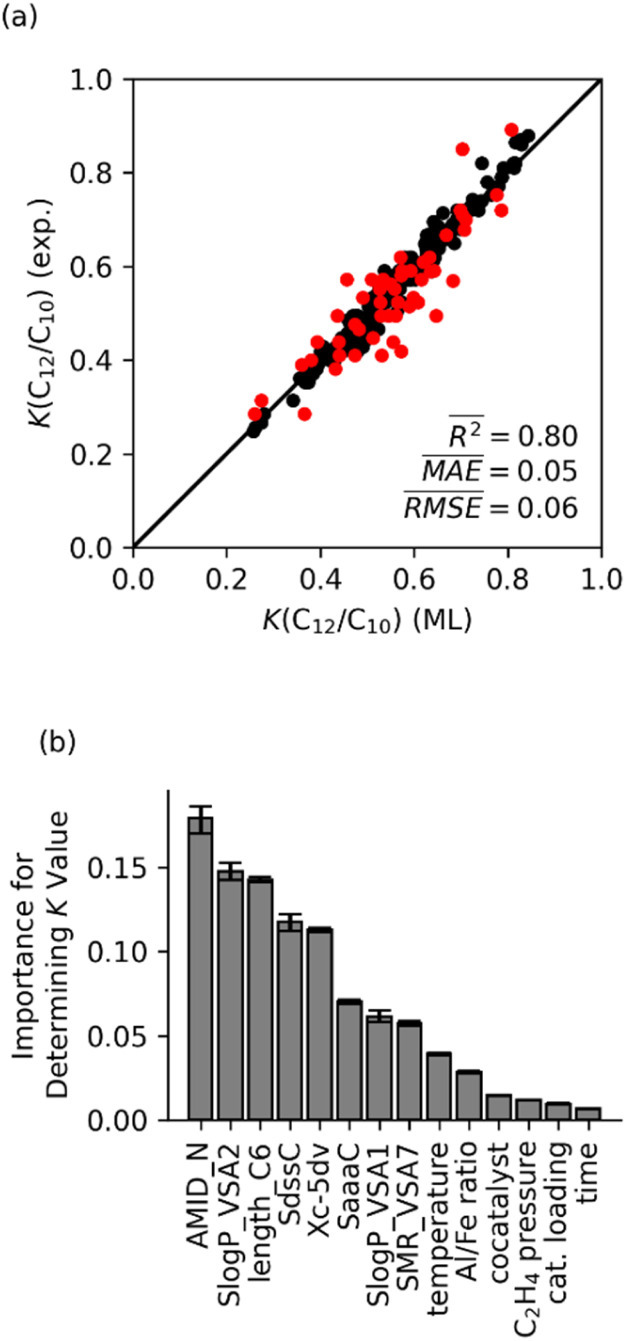
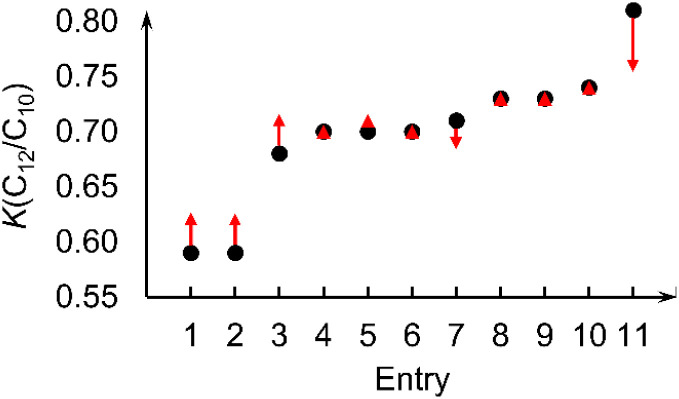
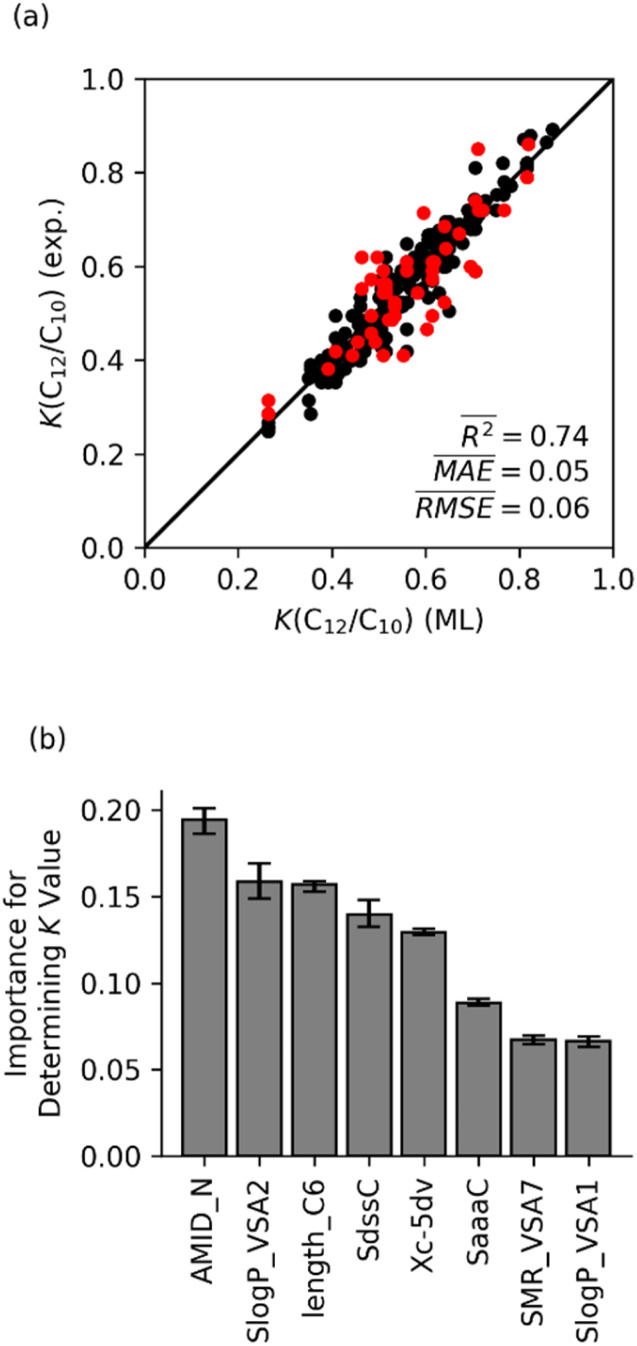
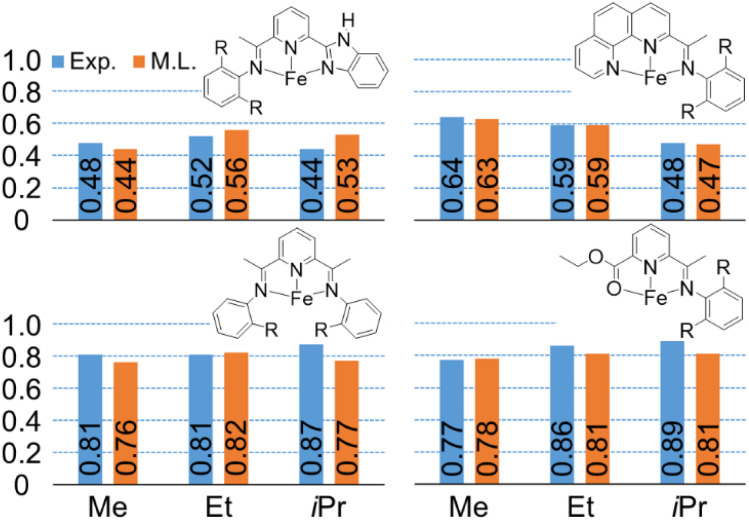
Linear α-olefins (1-alkenes) are critical comonomers for ethene copolymerization. A major impediment in the development of new homogeneous Fe catalysts for ethene oligomerization to produce comonomers and other important commercial products is the prediction of propagation termination rates that control the α-olefin distribution (, 1-butene through 1-decene), which is often referred to as a -value. Because the transition states for propagation termination are generally separated by less than a one kcal mol difference in energy, this selectivity cannot be accurately predicted by either DFT or wavefunction methods (even DLPNO-CCSD(T)). Therefore, we developed a sub-kcal mol accuracy machine learning model based on several hundred experimental selectivity values and straightforward 2D chemical and physical features that enables the prediction of α-olefin distribution -values. As part of our model, we developed a new feature that boosted the model performance. This machine learning model captures the effects of a broad range of ligand architectures and chemically nonintuitive trends in oligomerization selectivity. Our machine learning model was experimentally validated by prediction of a -value for a new Fe phosphaneyl-pyridinyl-quinoline catalyst followed by experimental measurement that showed precise agreement. In addition to quantitative predictions, we demonstrate how this machine learning model can provide qualitative catalyst design using proximity of pairs type analysis.
线性α-烯烃(1-烯烃)是乙烯共聚的关键共聚单体。开发用于乙烯齐聚以生产共聚单体和其他重要商业产品的新型均相铁催化剂的一个主要障碍是预测控制α-烯烃分布(从1-丁烯到1-癸烯)的链增长终止速率,这通常被称为α值。由于链增长终止的过渡态能量差通常小于1千卡/摩尔,这种选择性无法通过密度泛函理论(DFT)或波函数方法(甚至DLPNO-CCSD(T))准确预测。因此,我们基于数百个实验选择性值以及简单的二维化学和物理特征开发了一种亚千卡/摩尔精度的机器学习模型,该模型能够预测α-烯烃分布的α值。作为我们模型的一部分,我们开发了一种新特征,提高了模型性能。这种机器学习模型捕捉了广泛的配体结构的影响以及齐聚选择性中化学上非直观的趋势。我们的机器学习模型通过预测一种新型铁膦基吡啶基喹啉催化剂的α值并随后进行实验测量得到了验证,实验测量结果显示两者精确吻合。除了定量预测外,我们还展示了这种机器学习模型如何使用成对类型分析的接近度来提供定性的催化剂设计。