University of Tokyo, Department of Chemistry, Graduate School of Science 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
Chem Rev. 2024 Nov 13;124(21):12213-12241. doi: 10.1021/acs.chemrev.4c00422. Epub 2024 Oct 25.
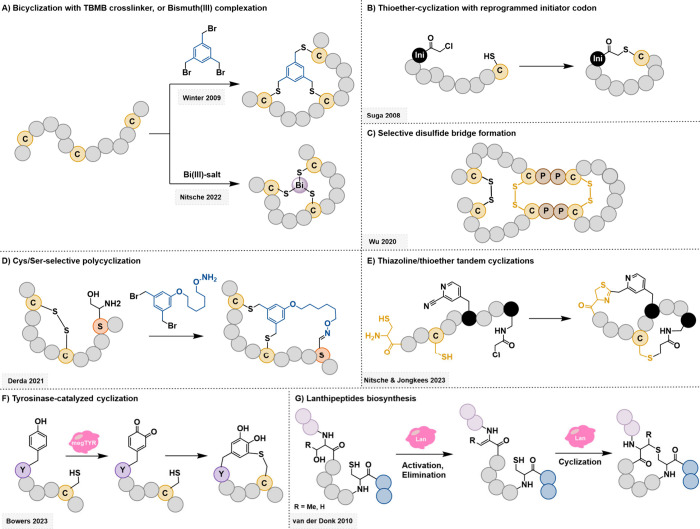
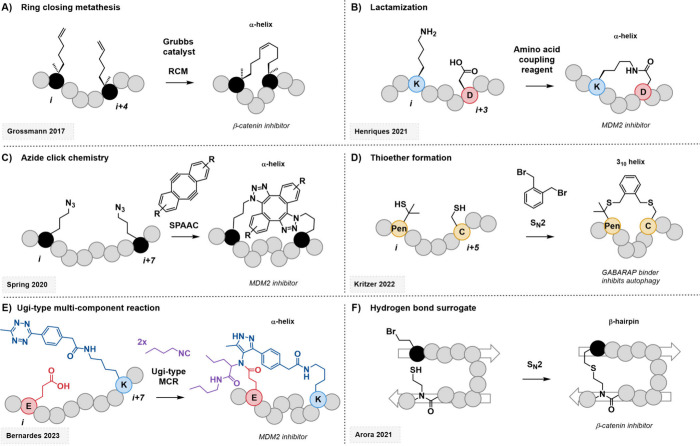
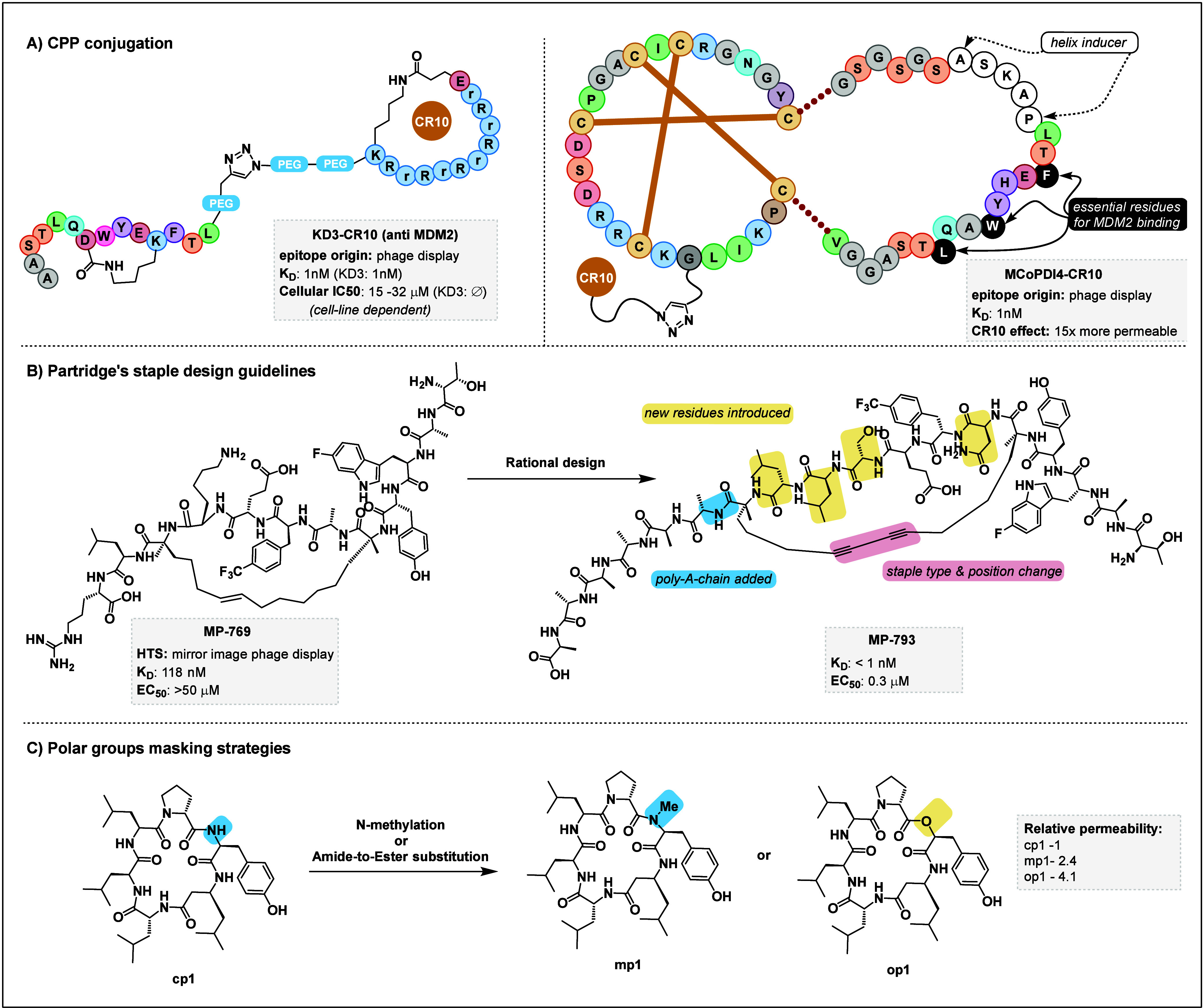
Technological advances and breakthrough developments in the pharmaceutical field are knocking at the door of the "undruggable" fortress with increasing insistence. Notably, the 21st century has seen the emergence of macrocyclic compounds, among which cyclic peptides are of particular interest. This new class of potential drug candidates occupies the vast chemical space between classic small-molecule drugs and larger protein-based therapeutics, such as antibodies. As research advances toward clinical targets that have long been considered inaccessible, macrocyclic peptides are well-suited to tackle these challenges in a post-rule of 5 pharmaceutical landscape. Facilitating their discovery is an arsenal of high-throughput screening methods that exploit massive randomized libraries of genetically encoded compounds. These techniques benefit from the incorporation of non-natural moieties, such as non- proteinogenic amino acids or stabilizing hydrocarbon staples. Exploiting these features for the strategic architectural design of macrocyclic peptides has the potential to tackle challenging targets such as protein-protein interactions, which have long resisted research efforts. This Review summarizes the basic principles and recent developments of the main high-throughput techniques for the discovery of macrocyclic peptides and focuses on their specific deployment for targeting undruggable space. A particular focus is placed on the development of new design guidelines and principles for the cyclization and structural stabilization of cyclic peptides and the resulting success stories achieved against well-known inaccessible drug targets.
技术进步和制药领域的突破性发展正越来越坚决地敲响“难成药”堡垒的大门。值得注意的是,21 世纪出现了大环化合物,其中环状肽尤其引人注目。这一新类潜在药物候选物占据了经典小分子药物和更大的基于蛋白质的治疗药物(如抗体)之间的广阔化学空间。随着研究向长期以来被认为难以触及的临床目标推进,大环肽非常适合在“后 5 规则”药物领域应对这些挑战。促进大环肽发现的是一系列高通量筛选方法,这些方法利用了大量遗传编码化合物的随机文库。这些技术受益于非天然部分的引入,例如非蛋白氨基酸或稳定的碳氢化合物支架。利用这些特性进行大环肽的战略架构设计有可能解决具有挑战性的目标,例如蛋白质-蛋白质相互作用,这些目标长期以来一直抵制研究工作。本文综述了大环肽发现的主要高通量技术的基本原理和最新进展,并重点介绍了它们在针对难成药空间方面的具体应用。特别关注的是环状肽的环化和结构稳定的新设计准则和原理的发展,以及针对知名难成药靶点所取得的成功案例。