Essoh Christiane, Hauck Yolande, Ouassa Timothée, Touré Daouda, Djatchi Richmond, Loukou Guillaume Yao, N'Guetta Simon-Pierre Assanvo, Vergnaud Gilles, Pourcel Christine
Département de Biochimie-Génétique, UFR des Sciences Biologiques, Université Peleforo Gon Coulibaly (UPGC), Korhogo BP 1328, Côte d'Ivoire.
Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Université Paris-Saclay, 91198 Gif-sur-Yvette, France.
Diagnostics (Basel). 2024 Oct 14;14(20):2284. doi: 10.3390/diagnostics14202284.
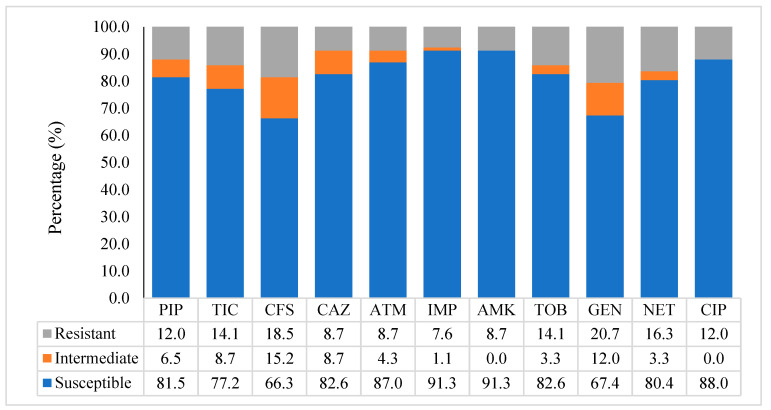
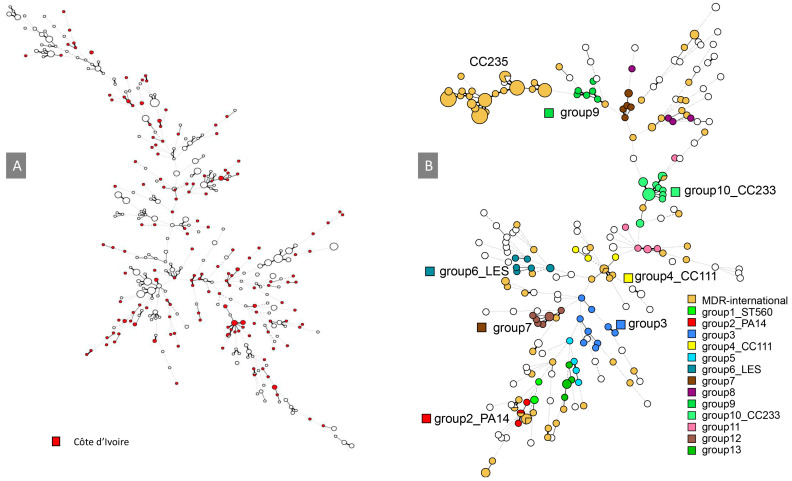
can cause community-acquired infections affecting various body sites. The present retrospective study investigated the genetic diversity of 173 isolates (166 clinical, 7 environmental) of collected from clinical pathology laboratories in Abidjan, Côte d'Ivoire (2001-2011). Multiple-Locus Variable Number of Tandem Repeats (VNTR) Analysis (MLVA) using 13 loci was applied to all isolates and compared to published MLVA data. The antibiotics status of the isolates was compiled when available and compared to published profiles. Among 95 isolates analyzed for their antibiotics status, 14 displayed concerning resistance profiles: five multidrug-resistant (MDR) and nine extensively drug-resistant (XDR). MLVA typing revealed a high genetic diversity (>130 genotypes), with many genotypes represented by a single strain. Notably, thirteen clusters (≥4 related isolates) were observed. Some clusters displayed close genetic relatedness to isolates from France, Korea, and well-studied strains (ST560, LES and PA14). Comparative analysis suggested the presence of international high-risk MDR clones (CC233, CC111) in Côte d'Ivoire. Importantly, MLVA clustering revealed a close relationship of CC235-MDR strains with a locally identified cluster (group 9). These findings support MLVA as a reliable and cost-effective tool for low-resource settings, allowing the selection of relevant strains for future whole genome sequence analyses. This approach can improve outbreak investigations and public health interventions aimed at curbing MDR transmission within hospitals and at the national level.
可引起影响身体各个部位的社区获得性感染。本回顾性研究调查了从科特迪瓦阿比让临床病理实验室收集的173株菌株(166株临床菌株,7株环境菌株)的遗传多样性(2001 - 2011年)。对所有分离株应用了使用13个位点的多位点可变串联重复序列(VNTR)分析(MLVA),并与已发表的MLVA数据进行比较。如有可用信息,会汇总分离株的抗生素耐药情况,并与已发表的概况进行比较。在分析其抗生素耐药情况的95株分离株中,14株显示出令人担忧的耐药谱:5株多重耐药(MDR)和9株广泛耐药(XDR)。MLVA分型显示出高度的遗传多样性(>130种基因型),许多基因型仅由单个菌株代表。值得注意的是,观察到13个聚类(≥4个相关分离株)。一些聚类与来自法国、韩国的分离株以及经过充分研究的菌株(ST560、LES和PA14)显示出密切的遗传相关性。比较分析表明科特迪瓦存在国际高危MDR克隆(CC233、CC111)。重要的是,MLVA聚类显示CC235 - MDR菌株与本地鉴定的一个聚类(第9组)关系密切。这些发现支持MLVA作为一种适用于资源匮乏环境的可靠且具有成本效益的工具,可用于选择相关菌株进行未来的全基因组序列分析。这种方法可以改善疫情调查以及旨在遏制医院内和国家层面MDR传播的公共卫生干预措施。