Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Department of Physics, Department of Anatomy and Cell Biology, University of Florida, Gainesville, FL, 32611, USA.
Nat Commun. 2024 Nov 2;15(1):9473. doi: 10.1038/s41467-024-53451-7.
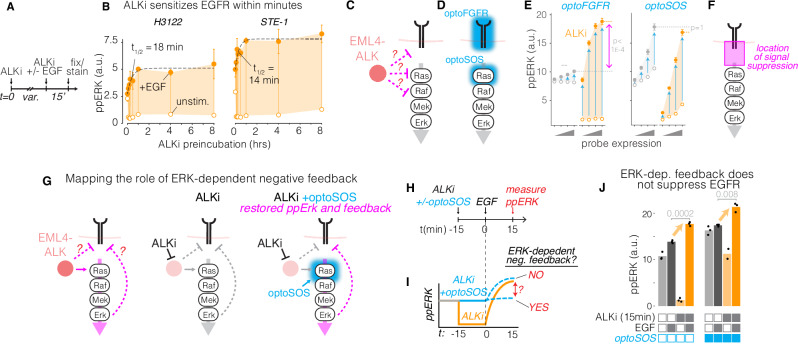
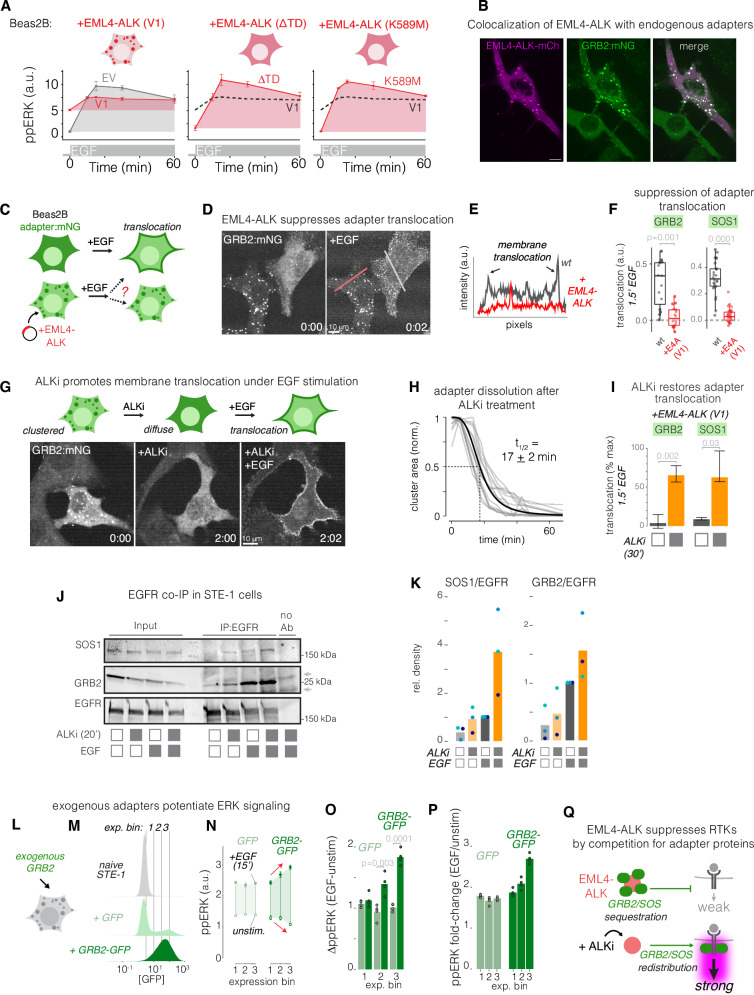
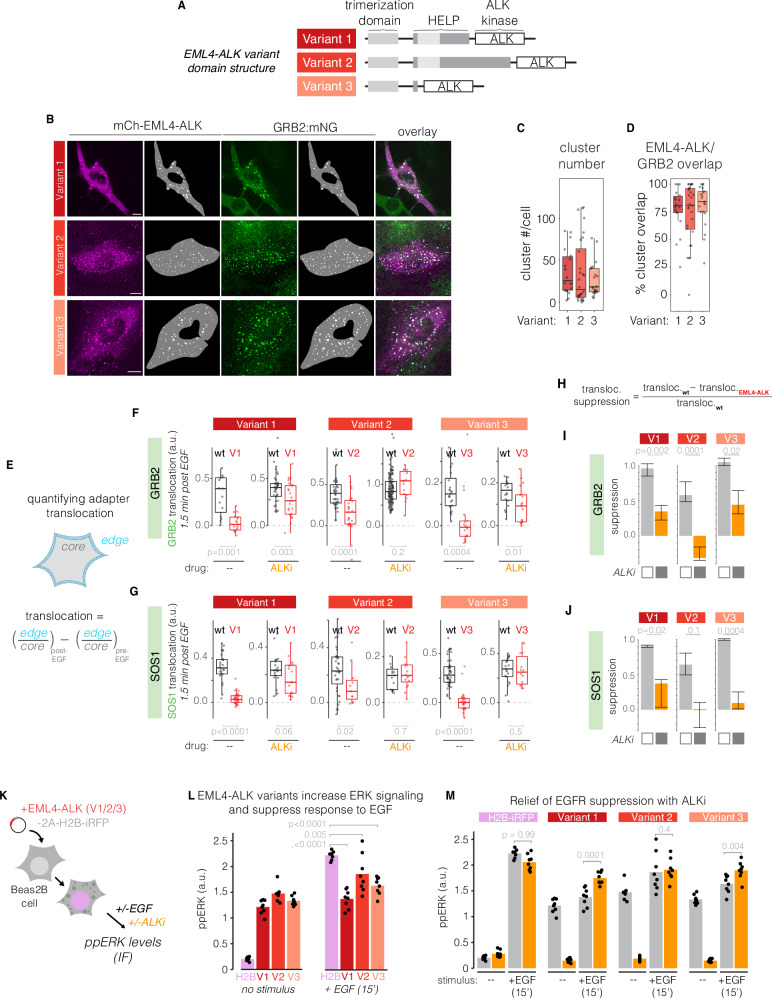
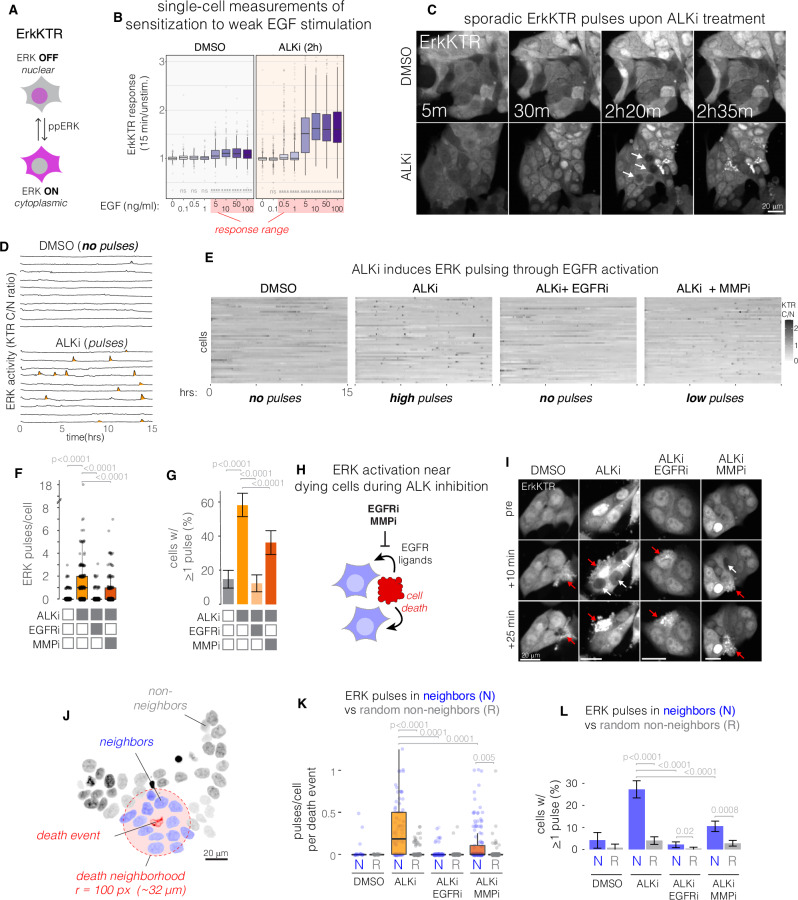
Drug resistance remains a challenge for targeted therapy of cancers driven by EML4-ALK and related fusion oncogenes. EML4-ALK forms cytoplasmic protein condensates, which result from networks of interactions between oncogene and adapter protein multimers. While these assemblies are associated with oncogenic signaling, their role in drug response is unclear. Here, we use optogenetics and live-cell imaging to find that EML4-ALK assemblies suppress transmembrane receptor tyrosine kinase (RTK) signaling by sequestering RTK adapter proteins including GRB2 and SOS1. Furthermore, ALK inhibition, while suppressing oncogenic signaling, simultaneously releases the sequestered adapters and thereby resensitizes RTK signaling. Resensitized RTKs promote rapid and pulsatile ERK reactivation that originates from paracrine ligands shed by dying cells. Reactivated ERK signaling promotes cell survival, which can be counteracted by combination therapies that block paracrine signaling. Our results identify a regulatory role for RTK fusion assemblies and uncover a mechanism of tolerance to targeted therapies.
药物耐药性仍然是由 EML4-ALK 和相关融合癌基因驱动的癌症靶向治疗的一个挑战。EML4-ALK 形成细胞质蛋白凝聚物,这是由于癌基因和衔接蛋白多聚体之间的相互作用网络导致的。虽然这些组装与致癌信号有关,但它们在药物反应中的作用尚不清楚。在这里,我们使用光遗传学和活细胞成像技术发现,EML4-ALK 组装通过隔离包括 GRB2 和 SOS1 在内的 RTK 衔接蛋白来抑制跨膜受体酪氨酸激酶(RTK)信号。此外,ALK 抑制虽然抑制了致癌信号,但同时也释放了被隔离的衔接蛋白,从而使 RTK 信号重新敏感化。重新敏感化的 RTKs 促进源自死亡细胞释放的旁分泌配体的快速和脉动的 ERK 再激活。再激活的 ERK 信号促进细胞存活,这可以通过阻断旁分泌信号的联合治疗来抵消。我们的研究结果确定了 RTK 融合组装的调节作用,并揭示了对靶向治疗产生耐受性的机制。