Centre of Excellence for Anaerobic Digestion, Department of Biology, University of York, Wentworth Way, Heslington, York, YO10 5DD, UK.
Microbiome. 2024 Nov 4;12(1):226. doi: 10.1186/s40168-024-01949-z.
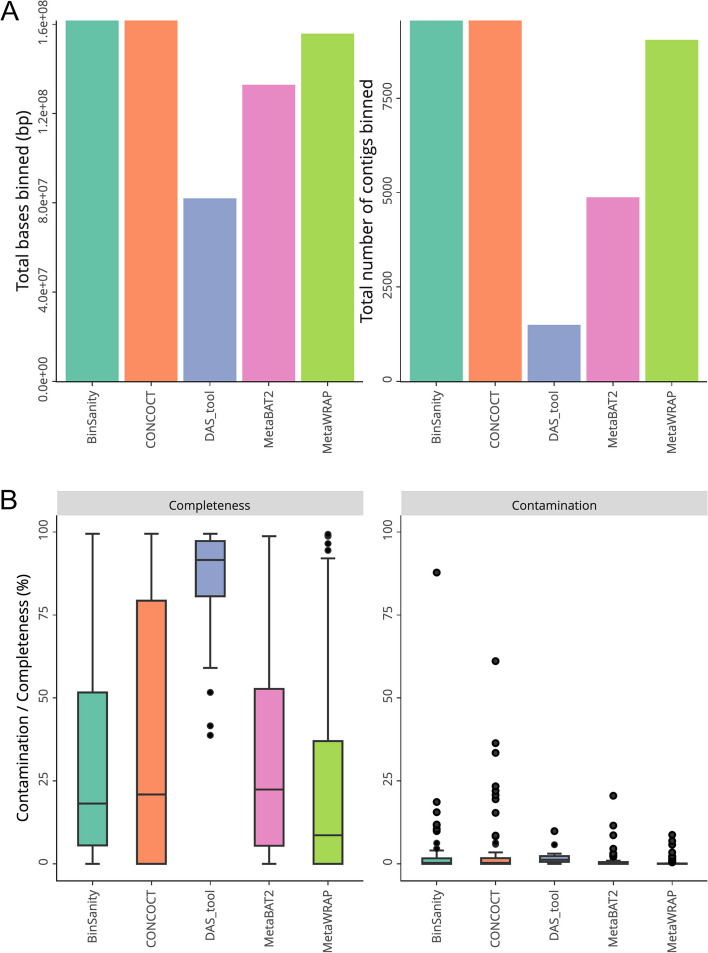
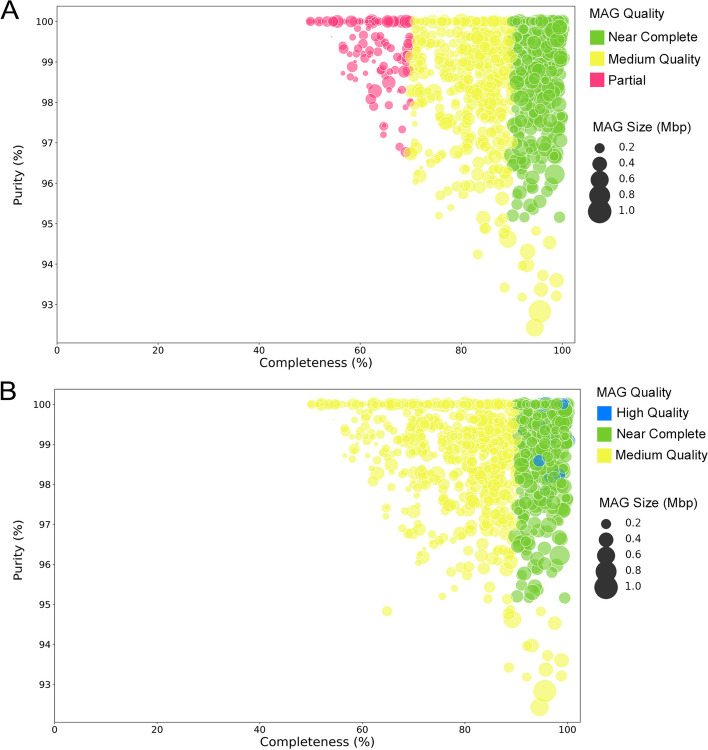
Metagenomics, the whole genome sequencing of microbial communities, has provided insight into complex ecosystems. It has facilitated the discovery of novel microorganisms, explained community interactions and found applications in various fields. Advances in high-throughput and third-generation sequencing technologies have further fuelled its popularity. Nevertheless, managing the vast data produced and addressing variable dataset quality remain ongoing challenges. Another challenge arises from the number of assembly and binning strategies used across studies. Comparing datasets and analysis tools is complex as it requires the quantitative assessment of metagenome quality. The inherent limitations of metagenomic sequencing, which often involves sequencing complex communities, mean community members are challenging to interrogate with traditional culturing methods leading to many lacking reference sequences. MIMAG standards aim to provide a method to assess metagenome quality for comparison but have not been widely adopted.
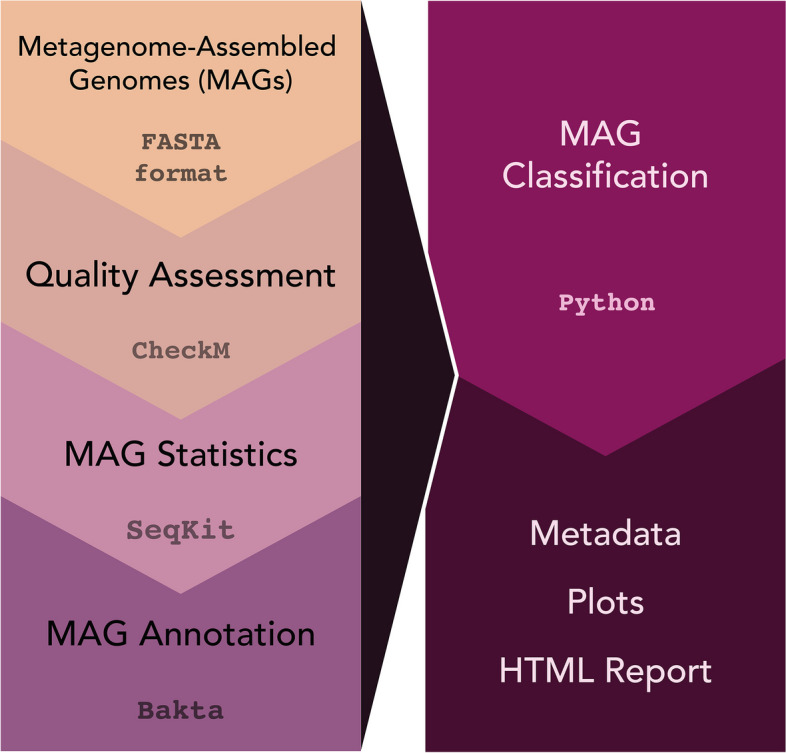
To address the need for simple and quick metagenome quality assignation, here we introduce the pipeline MAGqual (Metagenome-Assembled Genome qualifier) and demonstrate its effectiveness at determining metagenomic dataset quality in the context of the MIMAG standards.
The MAGqual pipeline offers an accessible way to evaluate metagenome quality and generate metadata on a large scale. MAGqual is built in Snakemake to ensure readability and scalability, and its open-source nature promotes accessibility, community development, and ease of updates. MAGqual is built in Snakemake, R, and Python and is available under the MIT license on GitHub at https://github.com/ac1513/MAGqual . Video Abstract.
宏基因组学,即对微生物群落的全基因组测序,为复杂生态系统提供了深入了解。它促进了新微生物的发现,解释了群落相互作用,并在各个领域得到了应用。高通量和第三代测序技术的进步进一步推动了其普及。然而,管理产生的大量数据和解决数据集质量变量仍然是持续存在的挑战。另一个挑战来自于研究中使用的组装和分类策略的数量。比较数据集和分析工具很复杂,因为它需要对宏基因组质量进行定量评估。宏基因组测序的固有局限性,通常涉及对复杂群落的测序,意味着用传统的培养方法很难对群落成员进行探究,导致许多缺乏参考序列。MIMAG 标准旨在提供一种评估宏基因组质量的方法以进行比较,但尚未得到广泛采用。
为了解决简单快速的宏基因组质量分配的需求,我们在这里引入了 MAGqual(宏基因组组装基因组质量评估)管道,并展示了它在 MIMAG 标准背景下确定宏基因组数据集质量的有效性。
MAGqual 管道提供了一种评估宏基因组质量的方法,并可大规模生成元数据。MAGqual 是在 Snakemake 中构建的,以确保可读性和可扩展性,其开源性质促进了可访问性、社区发展和轻松更新。MAGqual 构建在 Snakemake、R 和 Python 中,并在 GitHub 上以 MIT 许可证提供,网址为 https://github.com/ac1513/MAGqual。视频摘要。