Medical College, Guangxi University, Nanning 530004, China.
HIV/AIDS Clinical Treatment Center of Guangxi (Nanning) and The Fourth People's Hospital of Nanning, Nanning 530023, China.
Biosci Rep. 2024 Nov 27;44(11). doi: 10.1042/BSR20240595.
This study investigated the causal relationship between gut microbiota (GM), serum metabolome, and host transcriptome in the development of gout and hyperuricemia (HUA) using genome-wide association studies (GWAS) data and HUA mouse model experiments.
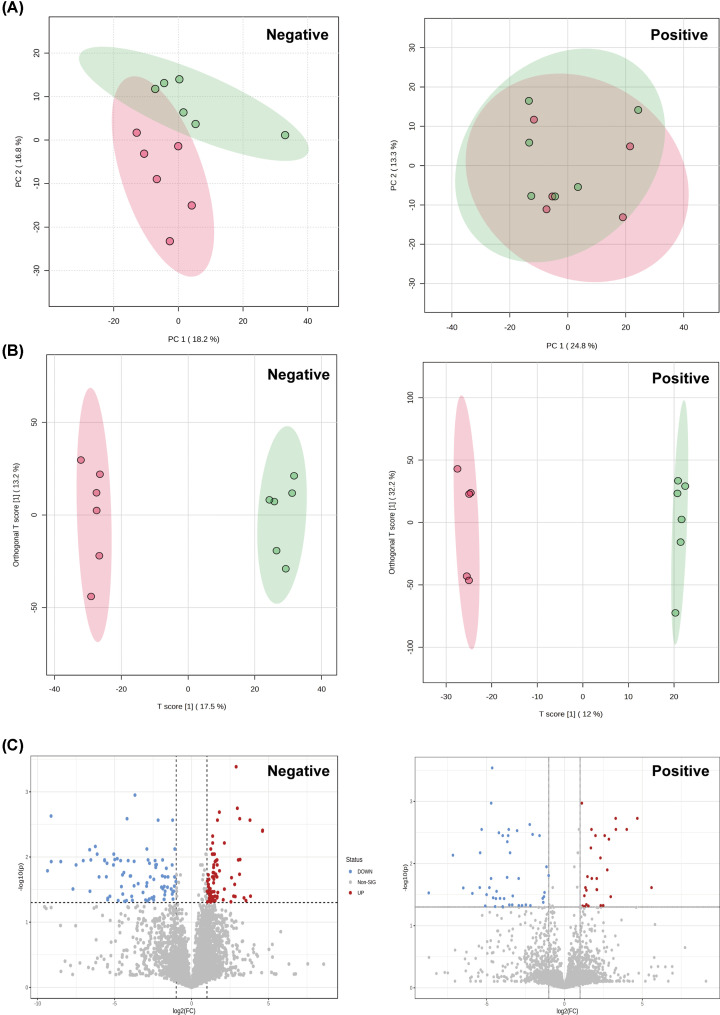
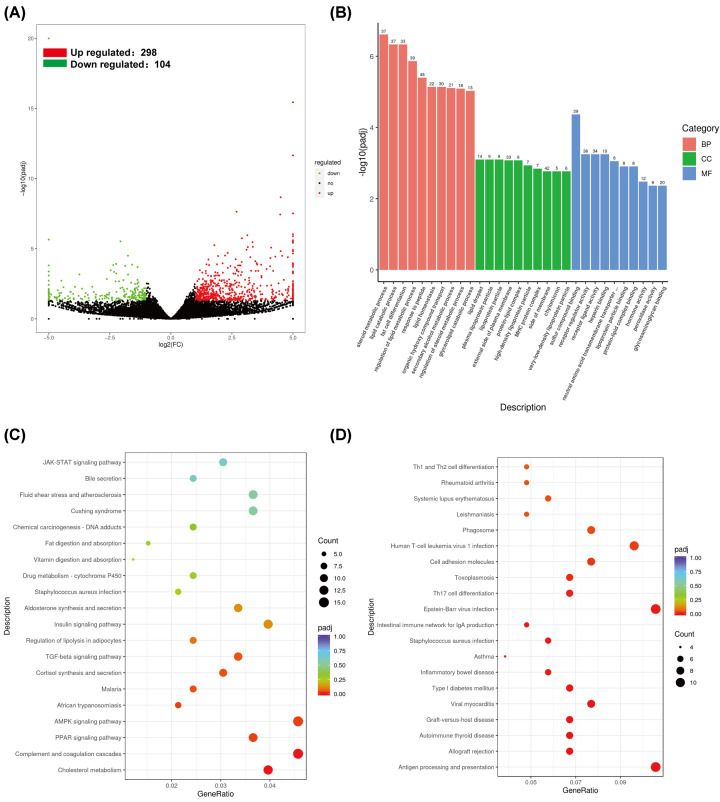
Mendelian randomization (MR) analysis of GWAS summary statistics was performed using an inverse variance weighted (IVW) approach to determine or predict the causal role of the GM on gout. The HUA mouse model was used to characterize changes in the gut microbiome, host metabolome, and host kidney transcriptome by integrating cecal 16S rRNA sequencing, untargeted serum metabolomics, and host mRNA sequencing.
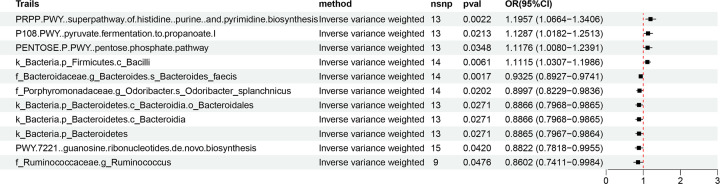
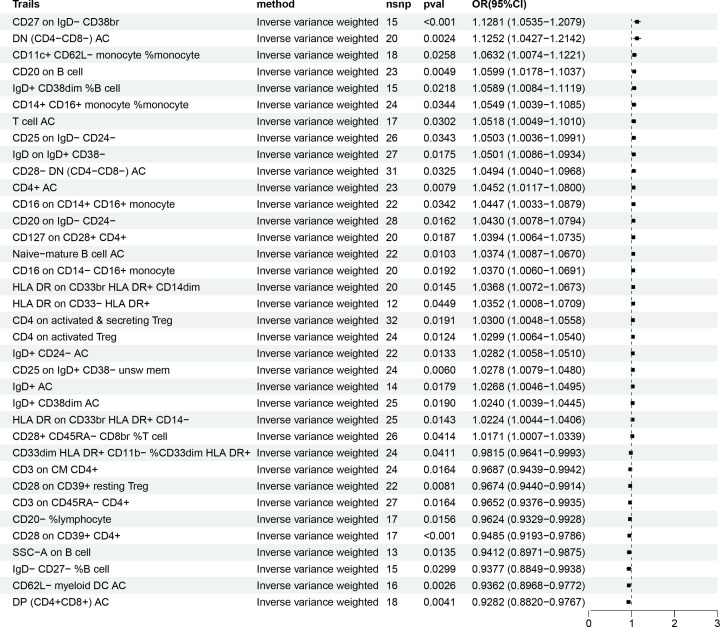
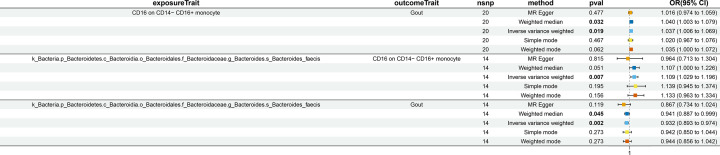
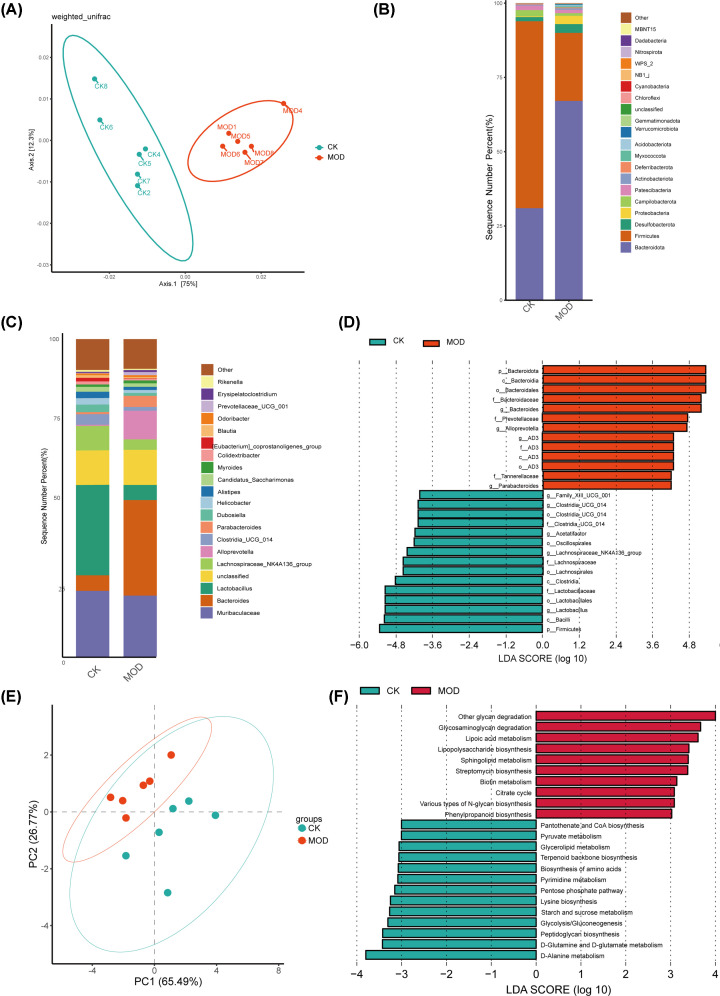
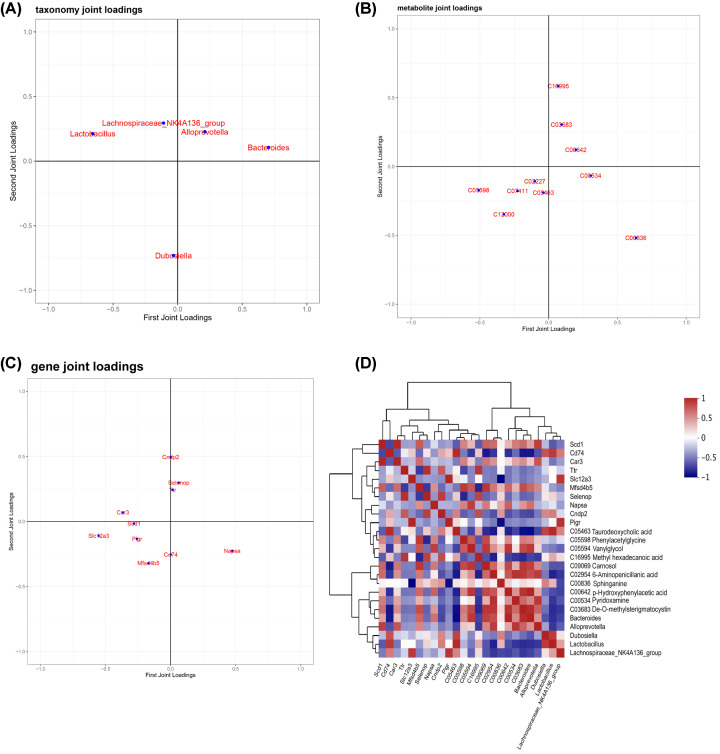
Our analysis demonstrated causal effects of seven GM taxa on gout, including genera of Ruminococcus, Odoribacter, and Bacteroides. Thirty eight immune cell traits were associated with gout. Dysbiosis of Dubosiella, Lactobacillus, Bacteroides, Alloprevotella, and Lachnospiraceae_NK4A136_group genera were associated with changes in the serum metabolites and kidney transcriptome of the HUA model mice. The changes in the gut microbiome of the HUA model mice correlated significantly with alterations in the levels of serum metabolites such as taurodeoxycholic acid, phenylacetylglycine, vanylglycol, methyl hexadecanoic acid, carnosol, 6-aminopenicillanic acid, sphinganine, p-hydroxyphenylacetic acid, pyridoxamine, and de-o-methylsterigmatocystin, and expression of kidney genes such as CNDP2, SELENOP, TTR, CAR3, SLC12A3, SCD1, PIGR, CD74, MFSD4B5, and NAPSA.
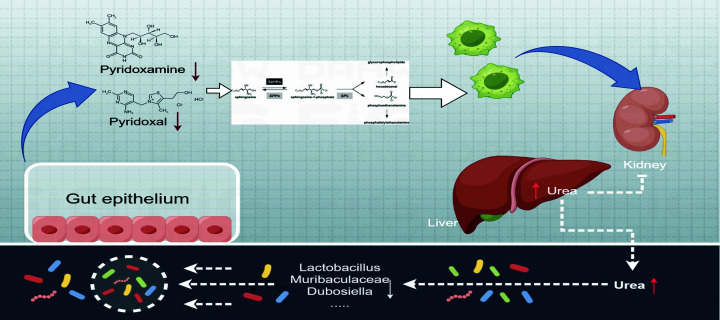
Our study demonstrated a causal relationship between GM, immune cells, and gout. HUA development involved alterations in the vitamin B6 metabolism because of GM dysbiosis that resulted in altered pyridoxamine and pyridoxal levels, dysregulated sphingolipid metabolism, and excessive inflammation.
本研究利用全基因组关联研究(GWAS)数据和高尿酸血症(HUA)小鼠模型实验,通过肠道微生物组(GM)、血清代谢组和宿主转录组的全基因组关联研究(GWAS)数据和 HUA 小鼠模型实验,研究 GM、血清代谢组和宿主转录组在痛风和高尿酸血症(HUA)发病中的因果关系。
采用逆方差加权(IVW)方法对 GWAS 汇总统计数据进行孟德尔随机化(MR)分析,以确定或预测 GM 对痛风的因果作用。采用 HUA 小鼠模型,通过整合盲肠 16S rRNA 测序、非靶向性血清代谢组学和宿主 mRNA 测序,研究肠道微生物组、宿主代谢组和宿主肾脏转录组的变化。
我们的分析表明,有 7 种 GM 分类群对痛风有因果影响,包括 Ruminococcus、Odoribacter 和 Bacteroides 属。38 种免疫细胞特征与痛风有关。Dubosiella、Lactobacillus、Bacteroides、Alloprevotella 和 Lachnospiraceae_NK4A136_group 属的失调与 HUA 模型小鼠的血清代谢物和肾脏转录组的变化有关。HUA 模型小鼠肠道微生物组的变化与血清代谢物水平的变化显著相关,如牛磺脱氧胆酸、苯乙酰甘氨酸、缬氨酰甘氨酸、甲基十六烷酸、卡诺醇、6-氨基青霉素酸、鞘氨醇、对羟基苯乙酸、吡哆胺和去-O-甲基表鬼笔环肽,以及肾脏基因的表达,如 CNDP2、SELENOP、TTR、CAR3、SLC12A3、SCD1、PIGR、CD74、MFSD4B5 和 NAPSA。
本研究表明 GM、免疫细胞与痛风之间存在因果关系。HUA 的发展涉及维生素 B6 代谢的改变,这是由于 GM 失调导致吡哆胺和吡哆醛水平改变、鞘脂代谢失调和过度炎症。