Peter Doherty Institute for Infection and Immunity, Department of Microbiology and Immunology, University of Melbourne, Melbourne, Australia.
Transmission, Infection, Diversification and Evolution Group, Max Planck Institute of Geoanthropology, Jena, Germany.
PLoS Comput Biol. 2024 Nov 6;20(11):e1012371. doi: 10.1371/journal.pcbi.1012371. eCollection 2024 Nov.
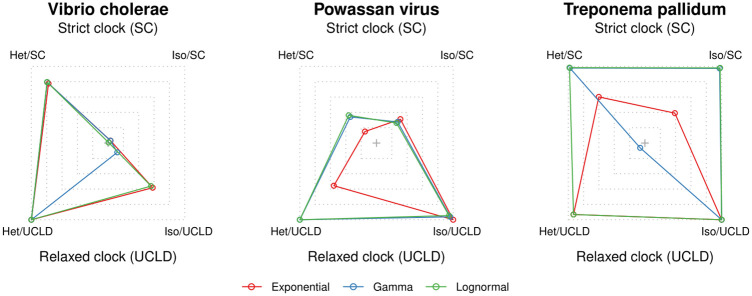
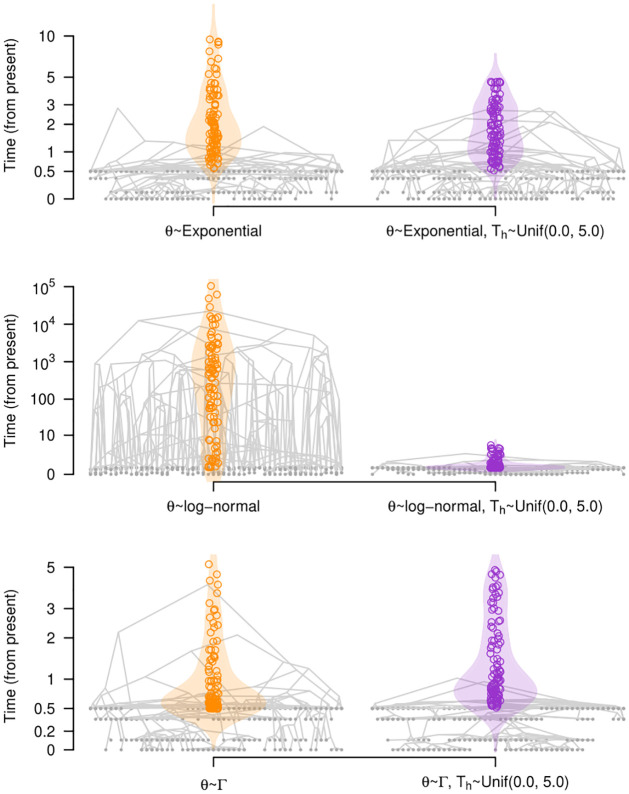
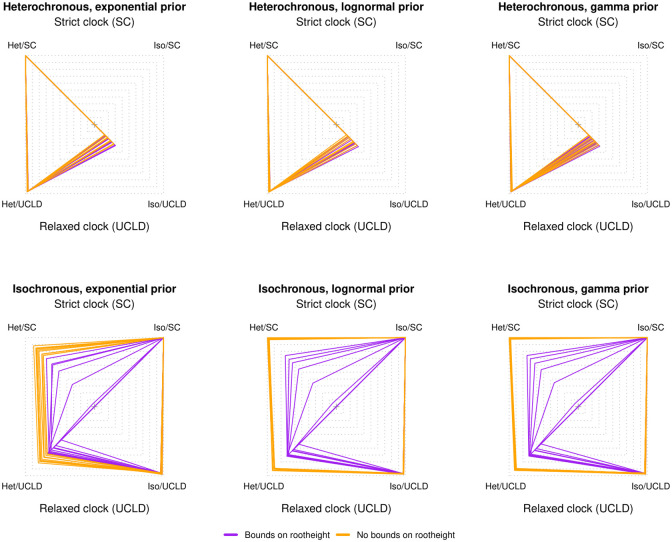
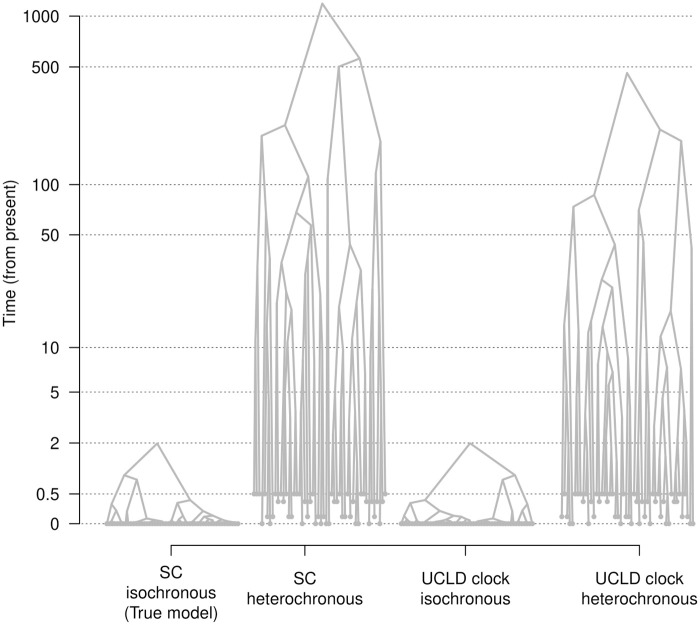
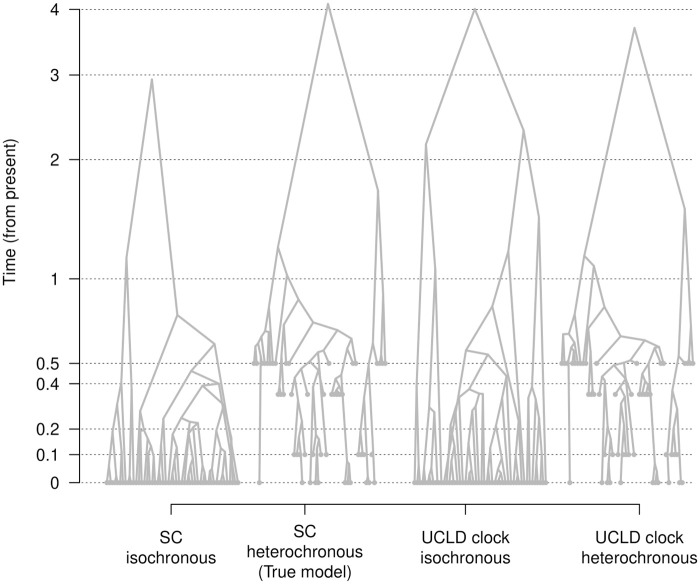
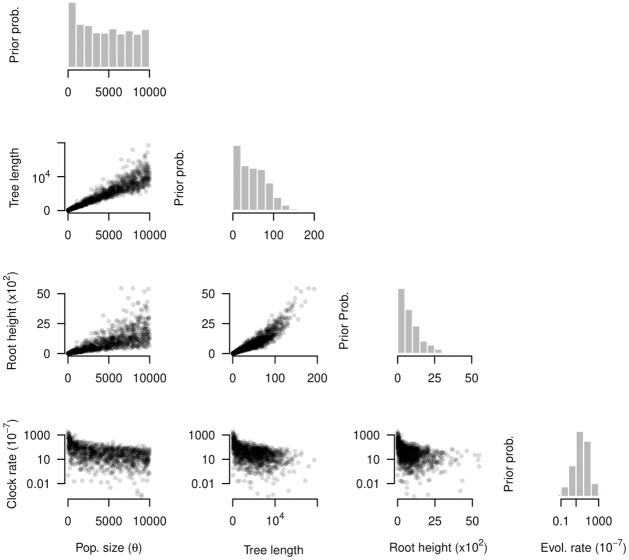
Our understanding of the evolution of many microbes has been revolutionised by the molecular clock, a statistical tool to infer evolutionary rates and timescales from analyses of biomolecular sequences. In all molecular clock models, evolutionary rates and times are jointly unidentifiable and 'calibration' information must therefore be used. For many organisms, sequences sampled at different time points can be employed for such calibration. Before attempting to do so, it is recommended to verify that the data carry sufficient information for molecular dating, a practice referred to as evaluation of temporal signal. Recently, a fully Bayesian approach, BETS (Bayesian Evaluation of Temporal Signal), was proposed to overcome known limitations of other commonly used techniques such as root-to-tip regression or date randomisation tests. BETS requires the specification of a full Bayesian phylogenetic model, posing several considerations for untangling the impact of model choice on the detection of temporal signal. Here, we aimed to (i) explore the effect of molecular clock model and tree prior specification on the results of BETS and (ii) provide guidelines for improving our confidence in molecular clock estimates. Using microbial molecular sequence data sets and simulation experiments, we assess the impact of the tree prior and its hyperparameters on the accuracy of temporal signal detection. In particular, highly informative priors that are inconsistent with the data can result in the incorrect detection of temporal signal. In consequence, we recommend: (i) using prior predictive simulations to determine whether the prior generates a reasonable expectation of parameters of interest, such as the evolutionary rate and age of the root node, (ii) conducting prior sensitivity analyses to assess the robustness of the posterior to the choice of prior, and (iii) selecting a molecular clock model that reasonably describes the evolutionary process.
我们对许多微生物进化的理解已经被分子钟所改变,这是一种从生物分子序列分析中推断进化率和时间尺度的统计工具。在所有分子钟模型中,进化率和时间是共同不可识别的,因此必须使用“校准”信息。对于许多生物体,可以使用在不同时间点采样的序列进行这种校准。在尝试这样做之前,建议验证数据是否携带足够的分子定年信息,这种做法被称为时间信号评估。最近,提出了一种完全贝叶斯方法 BETS(时间信号的贝叶斯评估),以克服其他常用技术(如根到尖端回归或日期随机化检验)的已知局限性。BETS 需要指定完整的贝叶斯系统发育模型,这对理清模型选择对检测时间信号的影响提出了一些考虑因素。在这里,我们旨在(i)探索分子钟模型和树先验规范对 BETS 结果的影响,以及(ii)为提高我们对分子钟估计的信心提供指导。使用微生物分子序列数据集和模拟实验,我们评估了树先验及其超参数对时间信号检测准确性的影响。特别是,与数据不一致的高度信息先验可能导致时间信号的错误检测。因此,我们建议:(i)使用先验预测模拟来确定先验是否生成了对感兴趣参数(例如进化率和根节点的年龄)的合理预期,(ii)进行先验敏感性分析以评估后验对先验选择的稳健性,以及(iii)选择合理描述进化过程的分子钟模型。