Angireddy Rajesh, Karisetty Bhanu Chandra, Katsura Kaitlin A, Díaz Abdias, Murali Svathi, Smith Sarina, Ohl Laura, Clark Kelly, Kossenkov Andrew V, Bhoj Elizabeth J K
Division of Human Genetics, Department of Pediatrics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, PA, USA.
The Wistar Institute, 3601 Spruce St., Philadelphia, PA 19104.
bioRxiv. 2024 Oct 31:2024.10.30.621078. doi: 10.1101/2024.10.30.621078.
TBCK syndrome is a rare fatal pediatric neurodegenerative disease caused by biallelic loss-of-function mutations in the gene. Previous studies by our lab and others have implicated mTOR, autophagy, lysosomes, and intracellular mRNA transport, however the exact primary pathologic mechanism is unknown. This gap has prevented the development of targeted therapies.
We employed a human neural progenitor cell line (NPC), ReNcell VM, which can differentiate into neurons and astrocytes, to understand the role of TBCK in mTORC1 activity and neuronal autophagy and cellular mechanisms of pathology. We used shRNA technology to knockdown TBCK in ReNcells.
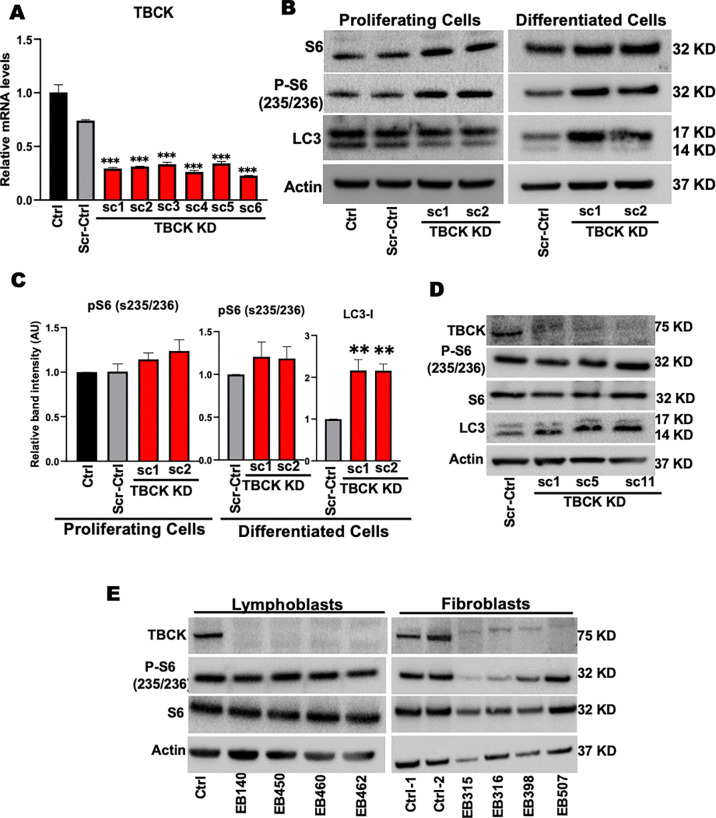
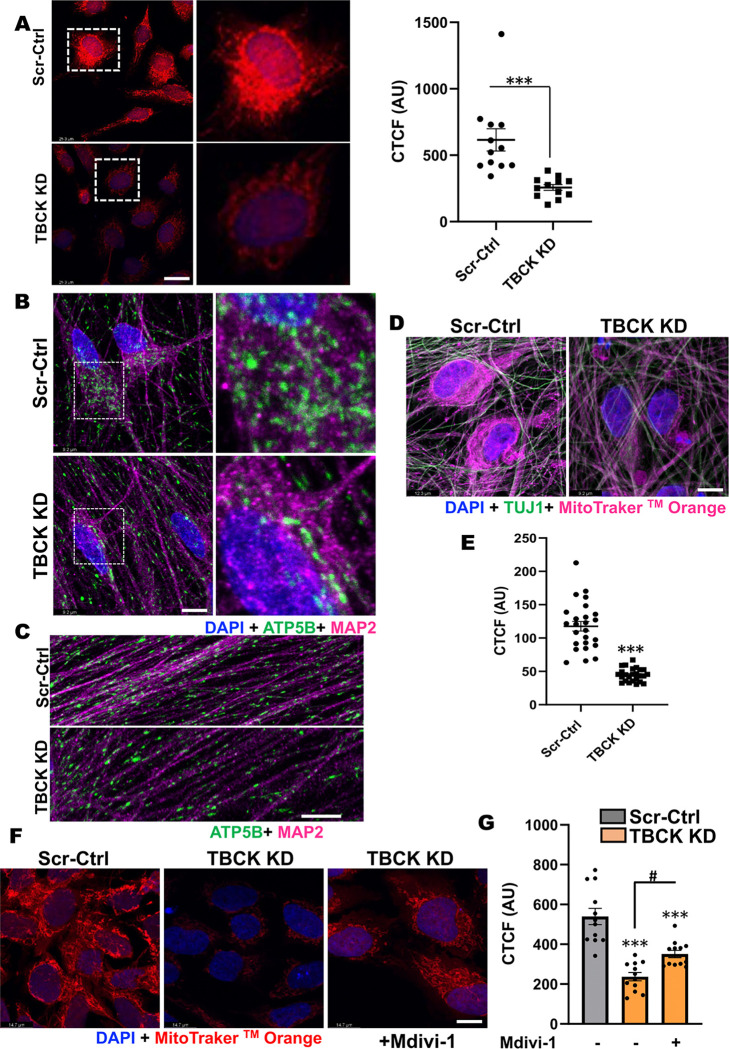
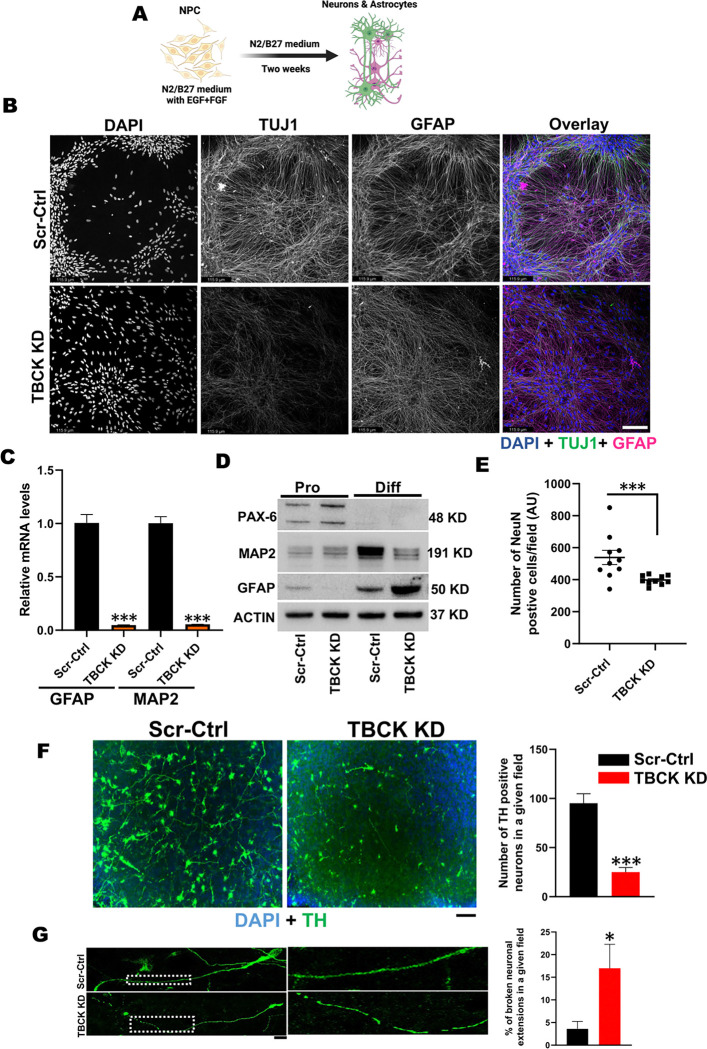
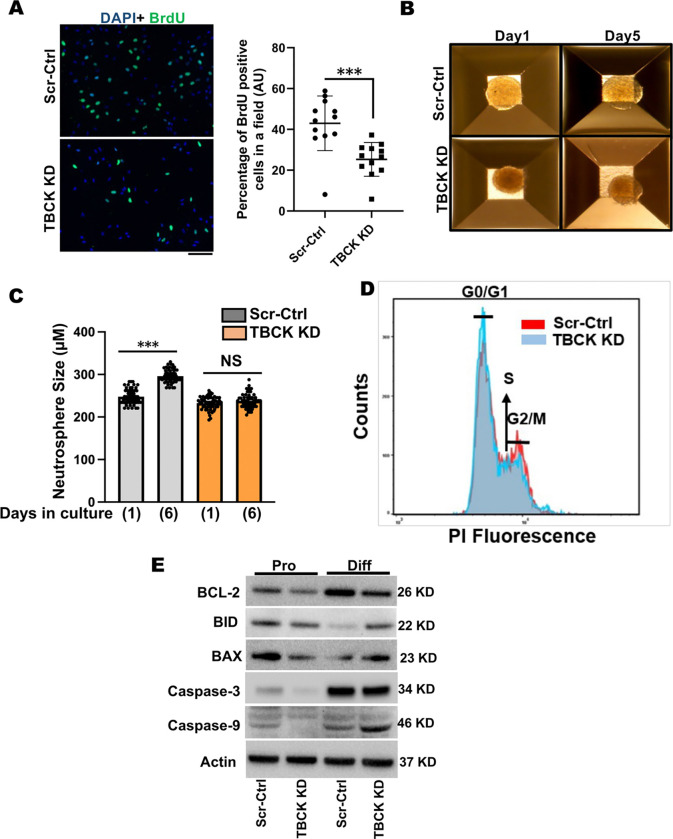
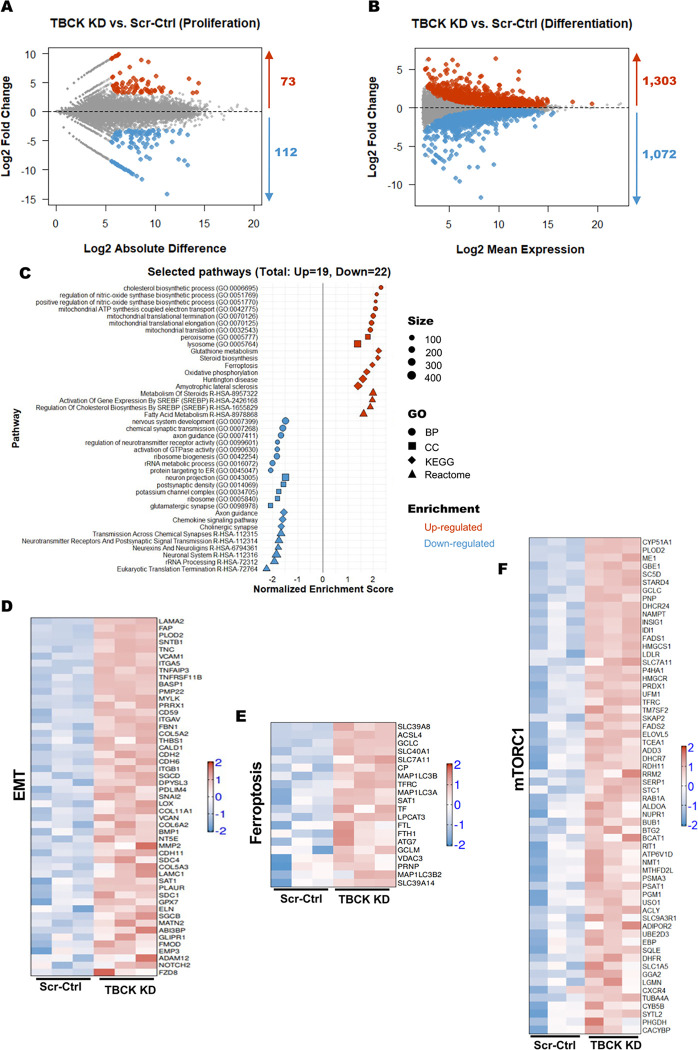
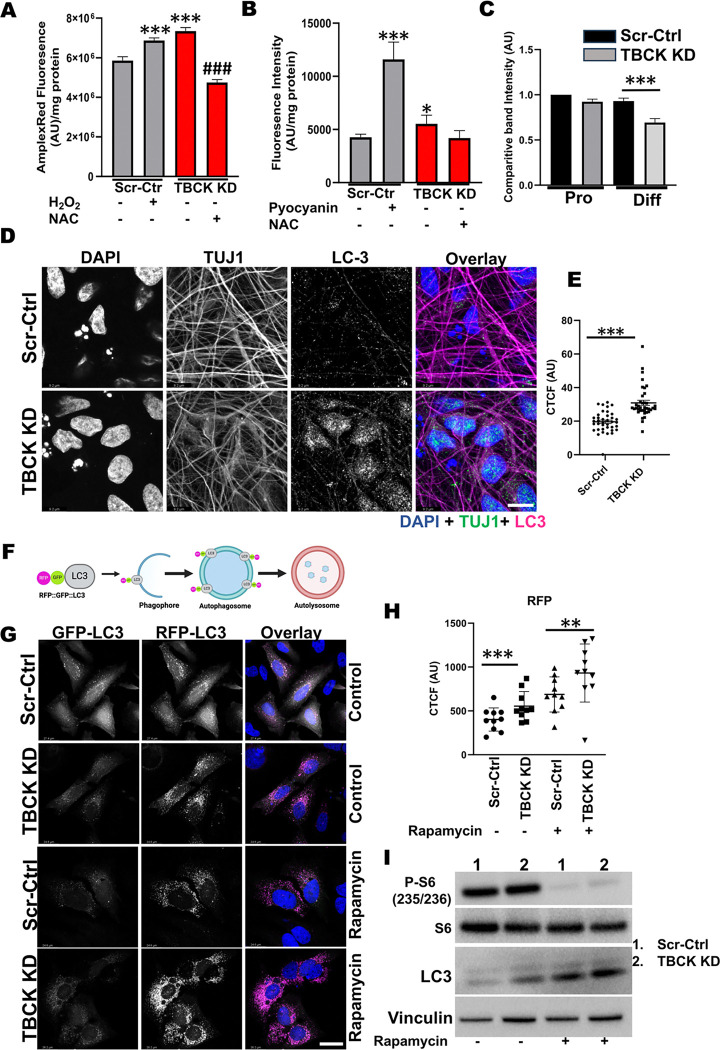
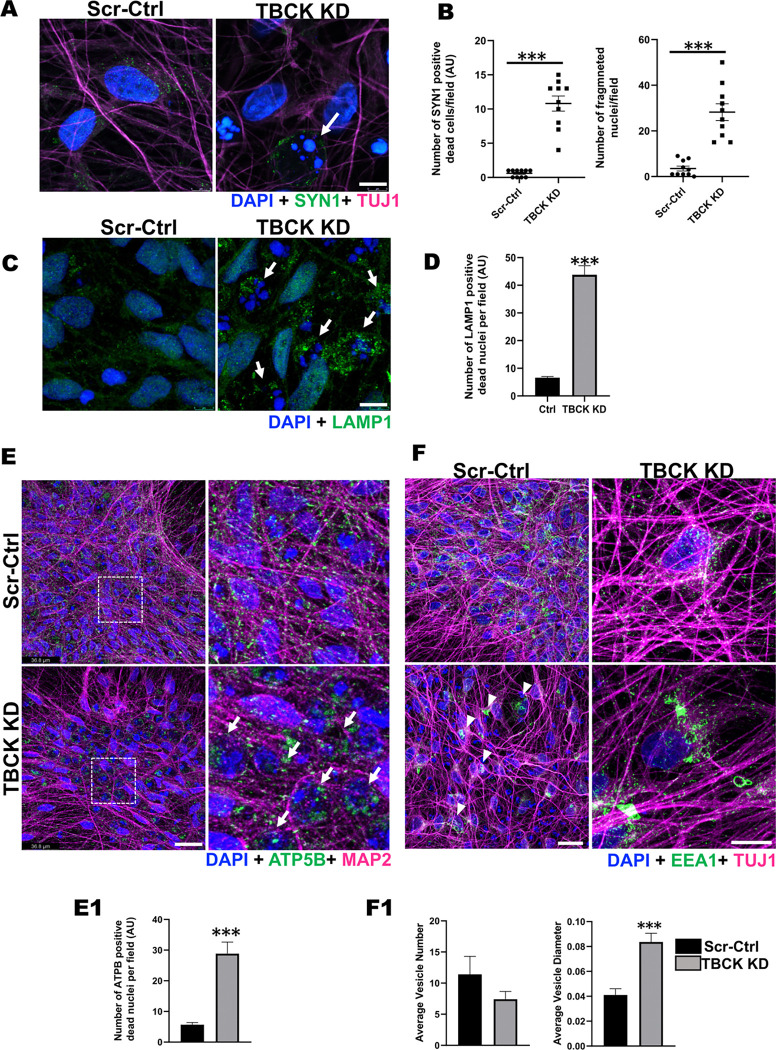
These data showed that loss of TBCK did not inhibit mTORC1 activity in neither NPC nor neurons. Additionally, analysis of eight patient-derived cells and TBCK knock down HeLa cells showed that mTORC1 inhibition is inconsistent across different patients and cell types. We showed that TBCK knockdown in ReNcells affected NPC differentiation to neurons and astrocytes. Specifically, differentiation defects are coupled to cell cycle defects in NPC and increased cell death during differentiation. RNAseq analysis indicated the downregulation of several different neurodevelopmental and differentiation pathways. We observed a higher number of LC3-positive vesicles in the soma and neurites of TBCK knockdown cells. Further, TBCK knockdown altered mitochondrial dynamics and membrane potential in NPC, neurons and astrocytes. We found partial mitochondrial rescue with the mitochondrial fission inhibitor mdivi-1.
This work outlines a new Human Cell Model for TBCK-related neurodegeneration and the essential role of mitochondrial health and partial rescue with mitochondrial fission inhibitor. This data, along with human neurons and astrocytes, illuminate mechanisms of neurodegeneration and provide a possible novel therapeutic avenue for affected patients.
TBCK综合征是一种由该基因双等位基因功能丧失突变引起的罕见致命性儿童神经退行性疾病。我们实验室及其他研究团队之前的研究表明mTOR、自噬、溶酶体和细胞内mRNA转运与之相关,但确切的主要病理机制尚不清楚。这一空白阻碍了靶向治疗的发展。
我们采用了一种人类神经祖细胞系(NPC)——ReNcell VM,它可以分化为神经元和星形胶质细胞,以了解TBCK在mTORC1活性、神经元自噬及病理细胞机制中的作用。我们使用shRNA技术在ReN细胞中敲低TBCK。
这些数据表明,TBCK缺失在NPC和神经元中均未抑制mTORC1活性。此外,对8个患者来源的细胞和TBCK敲低的HeLa细胞的分析表明,不同患者和细胞类型中mTORC1抑制情况不一致。我们发现,ReN细胞中TBCK敲低会影响NPC向神经元和星形胶质细胞的分化。具体而言,分化缺陷与NPC中的细胞周期缺陷相关,且分化过程中细胞死亡增加。RNA测序分析表明几种不同的神经发育和分化途径下调。我们在TBCK敲低细胞的胞体和神经突中观察到更多LC3阳性囊泡。此外,TBCK敲低改变了NPC、神经元和星形胶质细胞中的线粒体动力学和膜电位。我们发现线粒体裂变抑制剂mdivi-1可部分挽救线粒体。
这项工作概述了一种新的与TBCK相关神经退行性变的人类细胞模型,以及线粒体健康的重要作用和线粒体裂变抑制剂的部分挽救作用。这些数据与人类神经元和星形胶质细胞一起,阐明了神经退行性变的机制,并为受影响患者提供了一条可能的新治疗途径。