Cai Fei, Wei Yuehua, Kirchhofer Daniel, Chang Andrew, Zhang Yingnan
Departments of Biological Chemistry, Genentech, Inc., South San Francisco, California, United States of America.
DeepSeq.AI, Inc., San Francisco, California United States of America.
PLoS Comput Biol. 2024 Nov 18;20(11):e1012609. doi: 10.1371/journal.pcbi.1012609. eCollection 2024 Nov.
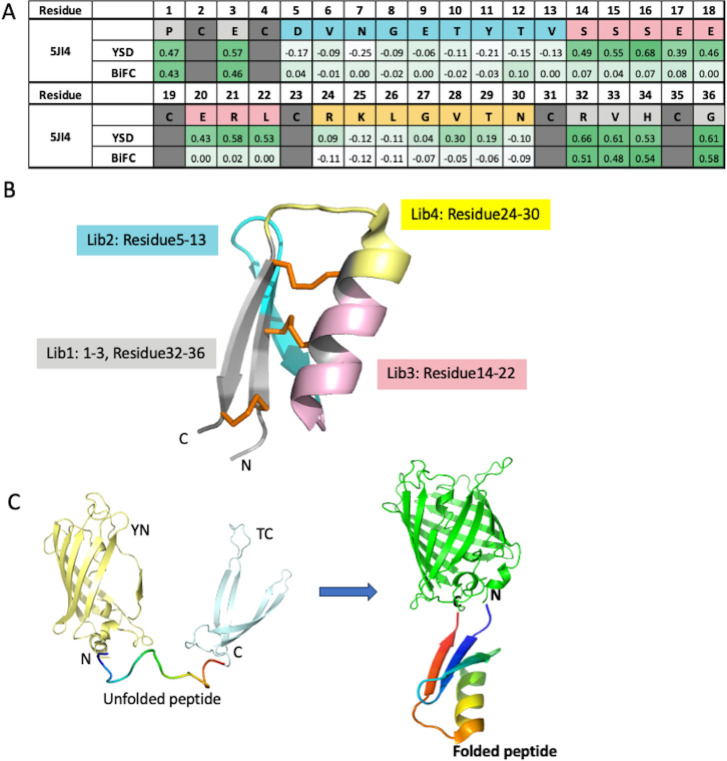
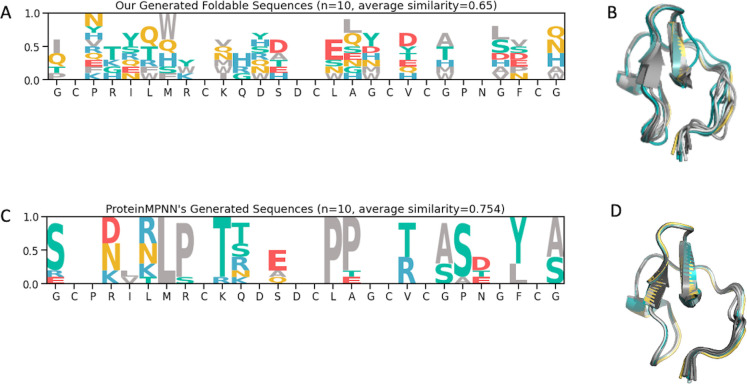
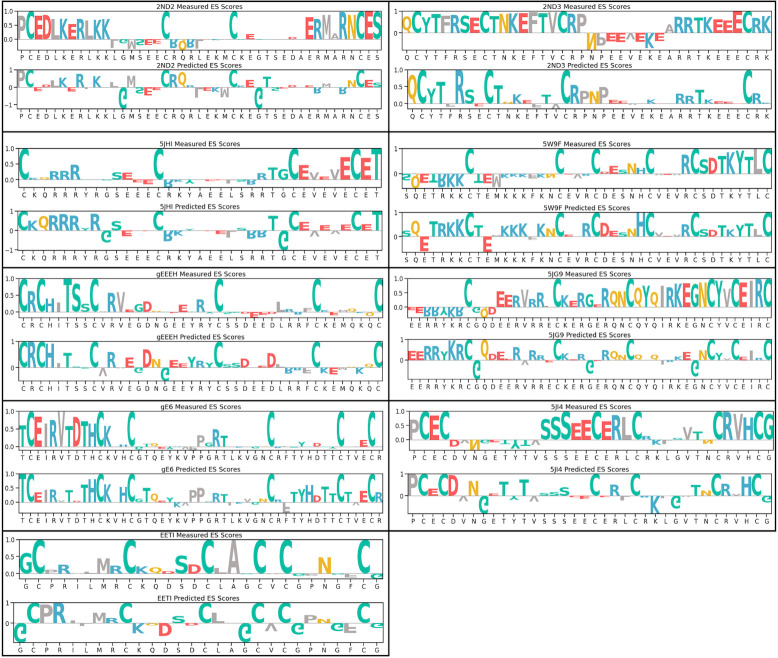
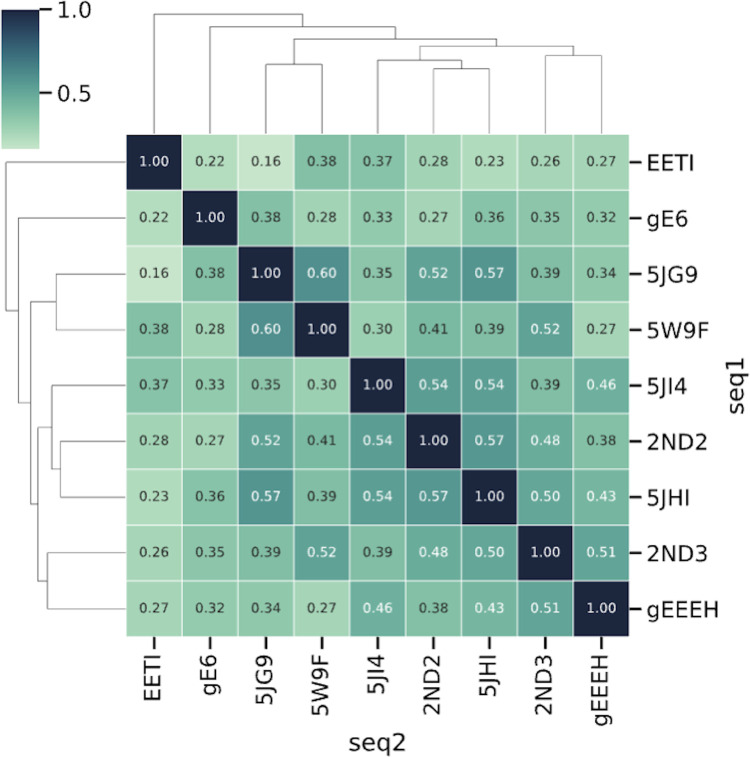
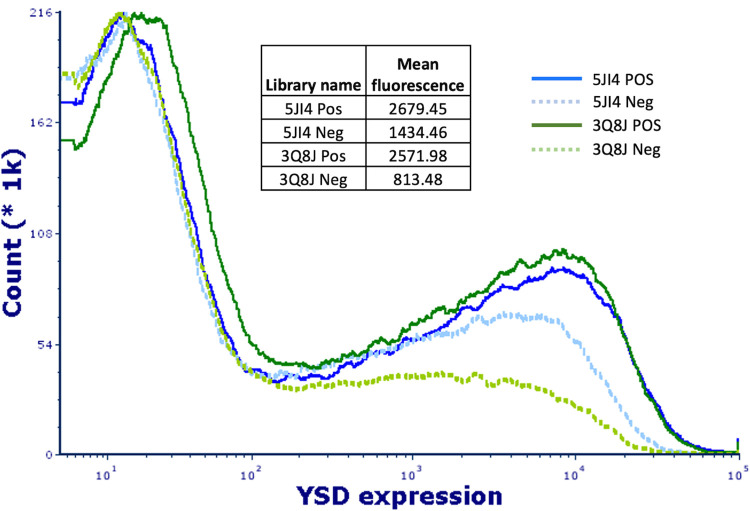
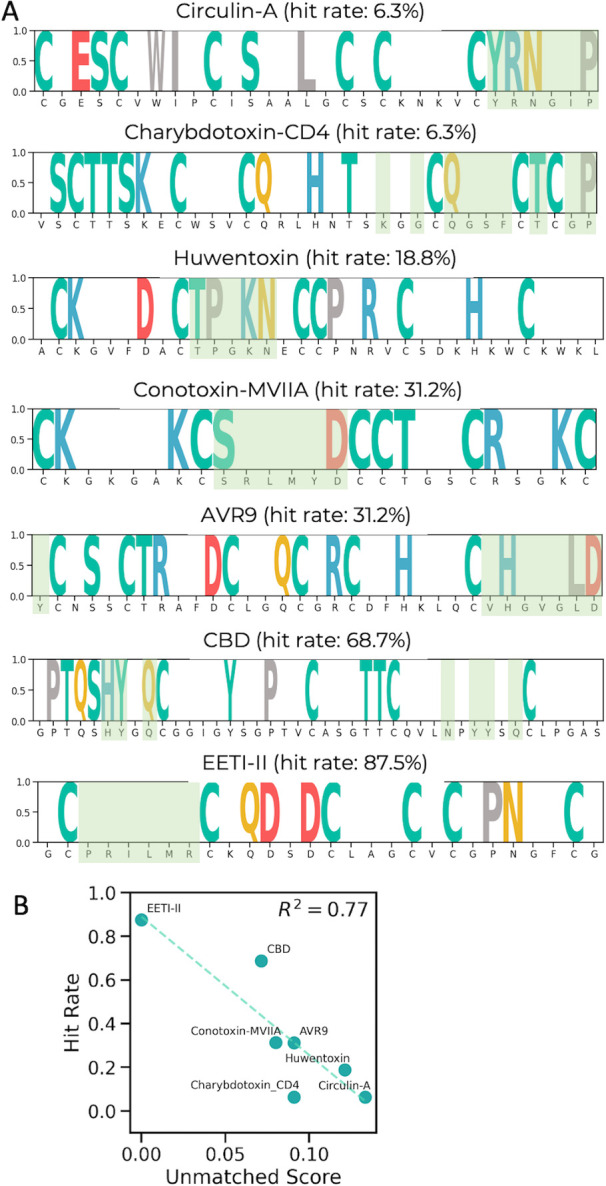
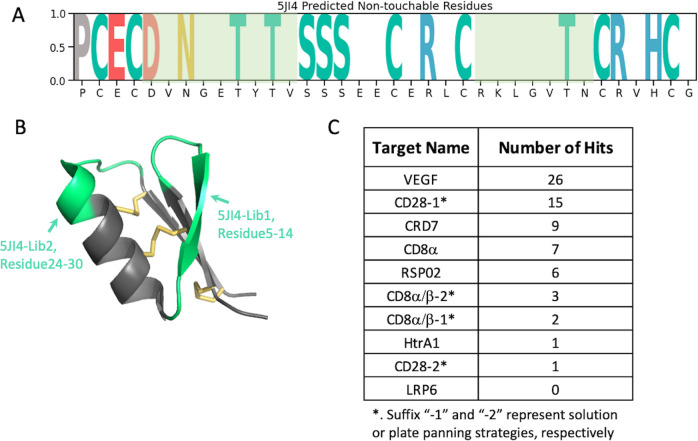
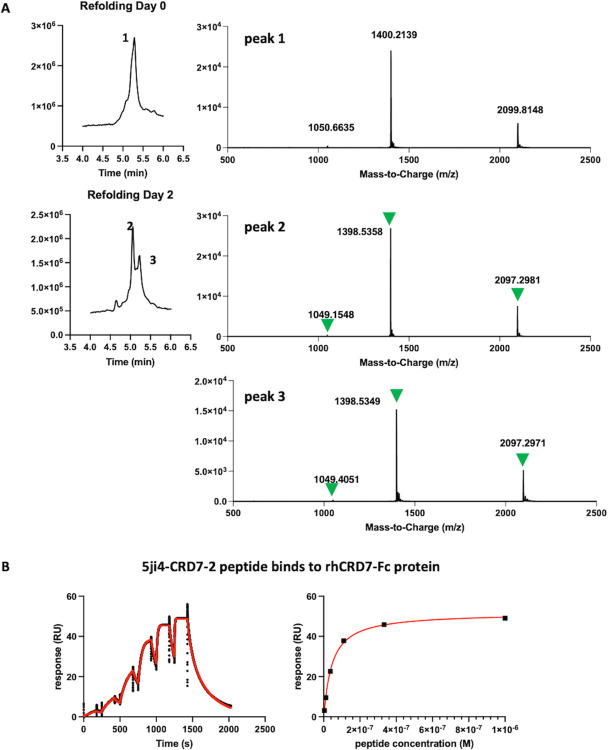
Peptides are an emerging modality for developing therapeutics that can either agonize or antagonize cellular pathways associated with disease, yet peptides often suffer from poor chemical and physical stability, which limits their potential. However, naturally occurring disulfide-constrained peptides (DCPs) and de novo designed Hyperstable Constrained Peptides (HCPs) exhibiting highly stable and drug-like scaffolds, making them attractive therapeutic modalities. Previously, we established a robust platform for discovering peptide therapeutics by utilizing multiple DCPs as scaffolds. However, we realized that those libraries could be further improved by considering the foldability of peptide scaffolds for library design. We hypothesized that specific sequence patterns within the peptide scaffolds played a crucial role in spontaneous folding into a stable topology, and thus, these sequences should not be subject to randomization in the original library design. Therefore, we developed a method for designing highly diverse DCP libraries while preserving the inherent foldability of each scaffold. To achieve this, we first generated a large-scale dataset from yeast surface display (YSD) combined with shotgun alanine scan experiments to train a machine-learning (ML) model based on techniques used for natural language understanding. Then we validated the ML model with experiments, showing that it is able to not only predict the foldability of peptides with high accuracy across a broad range of sequences but also pinpoint residues critical for foldability. Using the insights gained from the alanine scanning experiment as well as prediction model, we designed a new peptide library based on a de novo-designed HCP, which was optimized for enhanced folding efficiency. Subsequent panning trials using this library yielded promising hits having good folding properties. In summary, this work advances peptide or small protein domain library design practices. These findings could pave the way for the efficient development of peptide-based therapeutics in the future.
肽是一种新兴的治疗药物开发模式,它可以激活或拮抗与疾病相关的细胞通路,然而肽往往具有较差的化学和物理稳定性,这限制了它们的潜力。然而,天然存在的二硫键约束肽(DCP)和从头设计的超稳定约束肽(HCP)具有高度稳定且类似药物的支架结构,使其成为有吸引力的治疗模式。此前,我们利用多种DCP作为支架建立了一个强大的肽治疗药物发现平台。然而,我们意识到,通过在文库设计中考虑肽支架的可折叠性,这些文库可以得到进一步改进。我们假设肽支架内的特定序列模式在自发折叠成稳定拓扑结构中起着关键作用,因此,这些序列在原始文库设计中不应被随机化。因此,我们开发了一种在保留每个支架固有可折叠性的同时设计高度多样化DCP文库的方法。为了实现这一点,我们首先从酵母表面展示(YSD)结合鸟枪法丙氨酸扫描实验生成了一个大规模数据集,以基于用于自然语言理解的技术训练机器学习(ML)模型。然后我们通过实验验证了ML模型,表明它不仅能够在广泛的序列范围内高精度地预测肽的可折叠性,还能确定对可折叠性至关重要的残基。利用从丙氨酸扫描实验以及预测模型中获得的见解,我们基于一种从头设计的HCP设计了一个新的肽文库,该文库针对提高折叠效率进行了优化。随后使用该文库进行的淘选试验产生了具有良好折叠特性的有前景的命中序列。总之,这项工作推进了肽或小蛋白质结构域文库的设计实践。这些发现可能为未来基于肽的治疗药物的高效开发铺平道路。