Jurgens Sean J, Rämö Joel T, Kramarenko Daria R, Wijdeveld Leonoor F J M, Haas Jan, Chaffin Mark D, Garnier Sophie, Gaziano Liam, Weng Lu-Chen, Lipov Alex, Zheng Sean L, Henry Albert, Huffman Jennifer E, Challa Saketh, Rühle Frank, Verdugo Carmen Diaz, Krijger Juárez Christian, Kany Shinwan, van Orsouw Constance A, Biddinger Kiran, Poel Edwin, Elliott Amanda L, Wang Xin, Francis Catherine, Ruan Richard, Koyama Satoshi, Beekman Leander, Zimmerman Dominic S, Deleuze Jean-François, Villard Eric, Trégouët David-Alexandre, Isnard Richard, Boomsma Dorret I, de Geus Eco J C, Tadros Rafik, Pinto Yigal M, Wilde Arthur A M, Hottenga Jouke-Jan, Sinisalo Juha, Niiranen Teemu, Walsh Roddy, Schmidt Amand F, Choi Seung Hoan, Chang Kyong-Mi, Tsao Philip S, Matthews Paul M, Ware James S, Lumbers R Thomas, van der Crabben Saskia, Laukkanen Jari, Palotie Aarno, Amin Ahmad S, Charron Philippe, Meder Benjamin, Ellinor Patrick T, Daly Mark, Aragam Krishna G, Bezzina Connie R
Department of Experimental Cardiology, Amsterdam Cardiovascular Sciences, Heart Failure & Arrhythmias, Amsterdam UMC location, University of Amsterdam, Amsterdam, the Netherlands.
Cardiovascular Disease Initiative, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Nat Genet. 2024 Dec;56(12):2636-2645. doi: 10.1038/s41588-024-01975-5. Epub 2024 Nov 21.
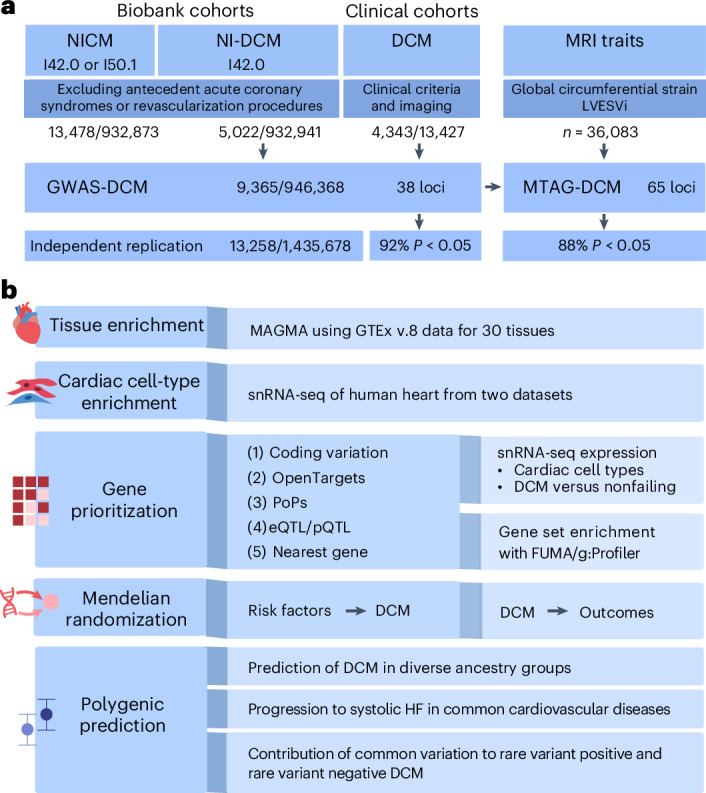
Dilated cardiomyopathy (DCM) is a heart muscle disease that represents an important cause of morbidity and mortality, yet causal mechanisms remain largely elusive. Here, we perform a large-scale genome-wide association study and multitrait analysis for DCM using 9,365 cases and 946,368 controls. We identify 70 genome-wide significant loci, which show broad replication in independent samples and map to 63 prioritized genes. Tissue, cell type and pathway enrichment analyses highlight the central role of the cardiomyocyte and contractile apparatus in DCM pathogenesis. Polygenic risk scores constructed from our genome-wide association study predict DCM across different ancestry groups, show differing contributions to DCM depending on rare pathogenic variant status and associate with systolic heart failure across various clinical settings. Mendelian randomization analyses reveal actionable potential causes of DCM, including higher bodyweight and higher systolic blood pressure. Our findings provide insights into the genetic architecture and mechanisms underlying DCM and myocardial function more broadly.
扩张型心肌病(DCM)是一种心肌疾病,是发病和死亡的重要原因,但其致病机制在很大程度上仍不清楚。在此,我们对9365例DCM病例和946368例对照进行了大规模全基因组关联研究和多性状分析。我们鉴定出70个全基因组显著位点,这些位点在独立样本中表现出广泛的重复性,并映射到63个优先基因。组织、细胞类型和通路富集分析突出了心肌细胞和收缩装置在DCM发病机制中的核心作用。根据我们的全基因组关联研究构建的多基因风险评分可预测不同祖先群体中的DCM,根据罕见致病变异状态对DCM有不同贡献,并在各种临床环境中与收缩性心力衰竭相关。孟德尔随机化分析揭示了DCM的可采取行动的潜在原因,包括体重增加和收缩压升高。我们的研究结果更广泛地为DCM和心肌功能的遗传结构及潜在机制提供了见解。