Shi Qian, Zhang Qimin, Shao Mingfu
Department of Computer Science and Engineering, School of Electrical Engineering and Computer Science, The Pennsylvania State University, University Park, PA 16802, USA.
Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA.
bioRxiv. 2024 Nov 6:2024.11.04.621958. doi: 10.1101/2024.11.04.621958.
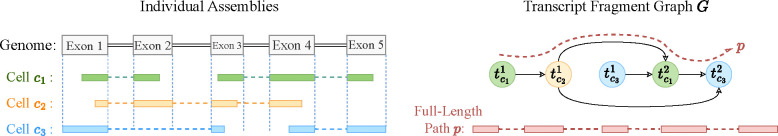
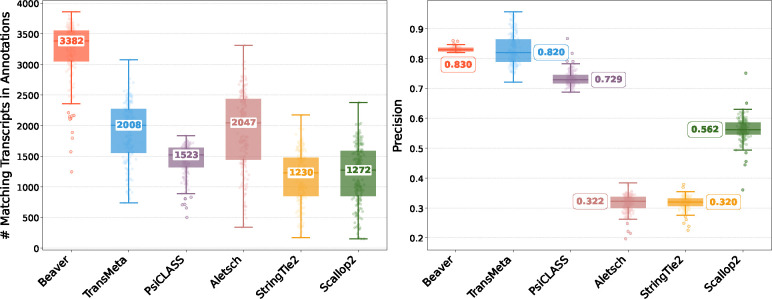
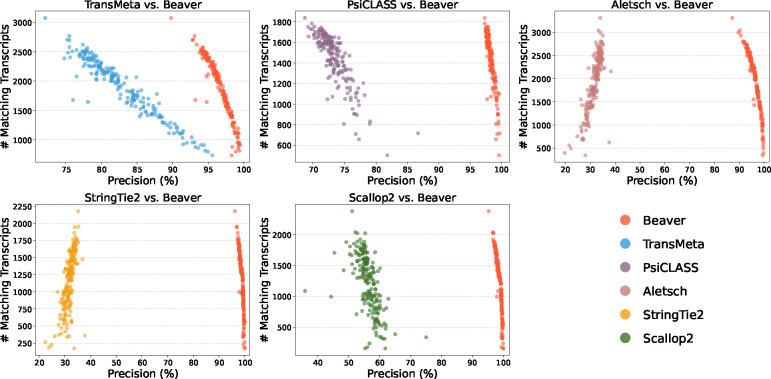
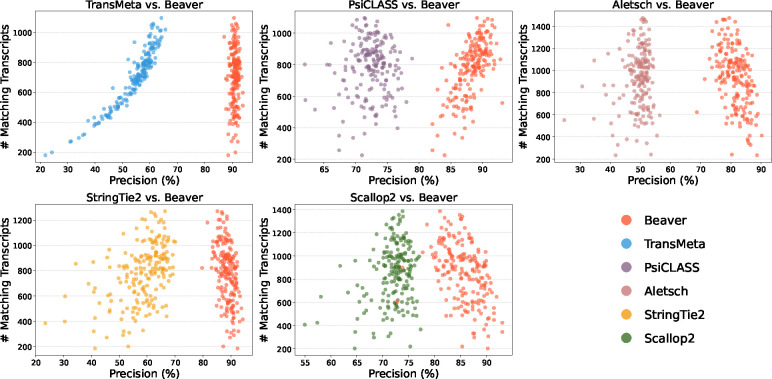
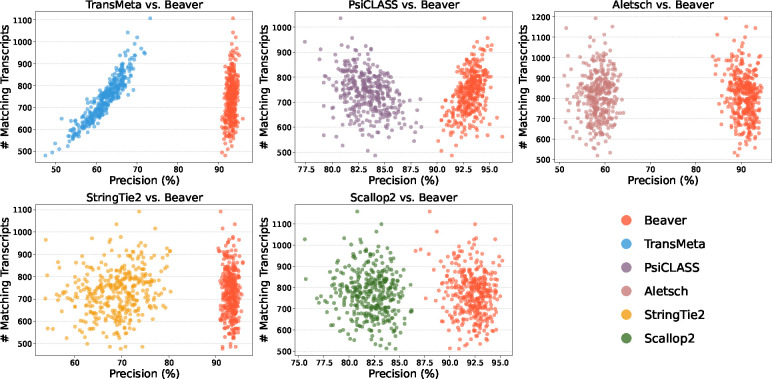
Emerging single-cell RNA sequencing techniques (scRNA-seq) has enabled the study of cellular transcriptome heterogeneity, yet accurate reconstruction of full-length transcripts at single-cell resolution remains challenging due to high dropout rates and sparse coverage. While meta-assembly approaches offer promising solutions by integrating information across multiple cells, current methods struggle to balance consensus assembly with cell-specific transcriptional signatures. Here, we present Beaver, a cell-specific transcript assembler designed for short-read scRNA-seq data. Beaver implements a transcript fragment graph to organize individual assemblies and designs an efficient dynamic programming algorithm that searches for candidate full-length transcripts from the graph. Beaver incorporates two random forest models trained on 51 meticulously engineered features that accurately estimate the likelihood of each candidate transcript being expressed in individual cells. Our experiments, performed using both real and simulated Smart-seq3 scRNA-seq data, firmly show that Beaver substantially outperforms existing meta-assemblers and single-sample assemblers. At the same level of sensitivity, Beaver achieved 32.0%-64.6%, 13.5%-36.6%, and 9.8%-36.3% higher precision in average compared to meta-assemblers Aletsch, TransMeta, and PsiCLASS, respectively, with similar improvements over single-sample assemblers Scallop2 (10.1%-43.6%) and StringTie2 (24.3%-67.0%). Beaver is freely available at https://github.com/Shao-Group/beaver. Scripts that reproduce the experimental results of this manuscript are available at https://github.com/Shao-Group/beaver-test.
新兴的单细胞RNA测序技术(scRNA-seq)使细胞转录组异质性研究成为可能,但由于高缺失率和稀疏覆盖,在单细胞分辨率下准确重建全长转录本仍然具有挑战性。虽然元组装方法通过整合多个细胞的信息提供了有前景的解决方案,但目前的方法难以在一致性组装和细胞特异性转录特征之间取得平衡。在这里,我们介绍了Beaver,一种为短读长scRNA-seq数据设计的细胞特异性转录本组装器。Beaver实现了一个转录本片段图来组织单个组装,并设计了一种高效的动态规划算法,从图中搜索候选全长转录本。Beaver纳入了两个基于51个精心设计的特征训练的随机森林模型,这些特征准确估计了每个候选转录本在单个细胞中表达的可能性。我们使用真实和模拟的Smart-seq3 scRNA-seq数据进行的实验有力地表明,Beaver明显优于现有的元组装器和单样本组装器。在相同的灵敏度水平下,与元组装器Aletsch、TransMeta和PsiCLASS相比,Beaver的平均精度分别提高了32.0%-64.6%、13.5%-36.6%和9.8%-36.3%,与单样本组装器Scallop2(10.1%-43.6%)和StringTie2(24.3%-67.0%)相比也有类似的提高。Beaver可在https://github.com/Shao-Group/beaver上免费获取。重现本手稿实验结果的脚本可在https://github.com/Shao-Group/beaver-test上获取。