Department of Preclinical Disciplines, Faculty of Medicine and Pharmacy, University of Oradea, 410081 Oradea, Romania.
Regional Center of Medical Genetics Bihor, County Emergency Clinical Hospital Oradea (Part of ERN-ITHACA), 410469 Oradea, Romania.
Medicina (Kaunas). 2024 Nov 20;60(11):1906. doi: 10.3390/medicina60111906.
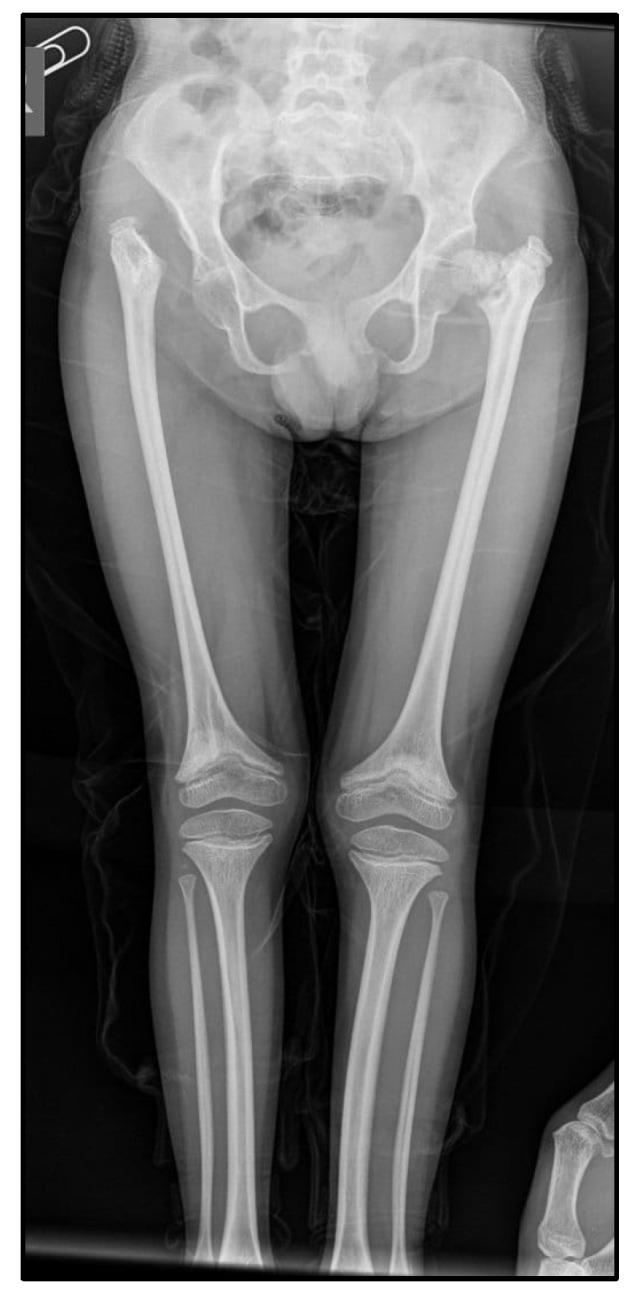
is a rare group of genetic conditions where individuals experience severe growth restriction, both in the womb and after birth. From as early as the fetal stage, those affected are significantly smaller than their peers. What makes PD distinct is its slow but steady growth pattern, resulting in proportionate dwarfism, where all parts of the body are equally shortened. Diagnosing and managing PD presents significant challenges due to its rarity and the wide range of clinical and genetic variability. The main conditions in this group include Seckel syndrome, Microcephalic Osteodysplastic Primordial Dwarfism (MOPD) types I/III, MOPD type II, Meier-Gorlin syndrome, and Silver-Russell syndrome (SRS). The first four-Seckel syndrome, MOPD types I/III, MOPD type II, and Meier-Gorlin syndrome-are associated with microcephaly, and together they are known as microcephalic PD. Given how uncommon PD is, establishing its exact incidence is difficult. It is estimated that about 4 million infants die within the first month of life, with 99% of these deaths occurring in the neonatal period. Accurately diagnosing PD requires meticulous evaluation, as it can be easily confused with other genetic disorders that also cause dwarfism. In this article, we present the case of a 10-year-old patient diagnosed with MOPD II, the most common and well-documented form of microcephalic PD. . Genetic analysis revealed a pathogenic variant in the gene ((c.1550dup, p.Gln518Alafs*7), alongside a deletion of exons 37-41. . This case sheds light on the clinical and genetic complexities of primordial dwarfism, underscoring the importance of timely and accurate diagnosis for effective patient care.
成骨不全-软骨发育不全-致死性发育不良综合征是一组罕见的遗传性疾病,患者在子宫内和出生后都会经历严重的生长受限。从胎儿期开始,受影响的个体就明显小于同龄人。成骨不全-软骨发育不全-致死性发育不良综合征的独特之处在于其缓慢而稳定的生长模式,导致成比例的侏儒症,即身体所有部位都同等缩短。由于其罕见性和广泛的临床及遗传变异性,诊断和管理成骨不全-软骨发育不全-致死性发育不良综合征都极具挑战性。该综合征主要包括 Seckel 综合征、小头畸形性骨发育不良性矮小症(MOPD)Ⅰ/Ⅲ型、MOPD Ⅱ型、Meier-Gorlin 综合征和 Silver-Russell 综合征(SRS)。前四种——Seckel 综合征、MOPDⅠ/Ⅲ型、MOPD Ⅱ型和 Meier-Gorlin 综合征——与小头症相关,统称为小头性成骨不全-软骨发育不全-致死性发育不良综合征。由于成骨不全-软骨发育不全-致死性发育不良综合征非常罕见,因此很难确定其确切的发病率。据估计,约有 400 万婴儿在出生后的第一个月内死亡,其中 99%的死亡发生在新生儿期。准确诊断成骨不全-软骨发育不全-致死性发育不良综合征需要进行细致的评估,因为它很容易与其他也导致侏儒症的遗传性疾病混淆。在本文中,我们介绍了一位 10 岁患者的病例,该患者被诊断为 MOPD Ⅱ型,这是最常见且记录最完善的小头性成骨不全-软骨发育不全-致死性发育不良综合征。基因分析显示,该患者存在基因(c.1550dup,p.Gln518Alafs*7)的致病性变异,同时还存在外显子 37-41 的缺失。该病例揭示了原发性侏儒症的临床和遗传复杂性,强调了及时准确诊断对于有效患者护理的重要性。