Tae Han-Shen, Hung Andrew, Clark Richard J, Adams David J
Molecular Horizons, Faculty of Science, Medicine and Health, University of Wollongong, Wollongong, Australia.
School of Science, RMIT University, Melbourne, Australia.
J Biol Chem. 2025 Jan;301(1):108017. doi: 10.1016/j.jbc.2024.108017. Epub 2024 Nov 26.
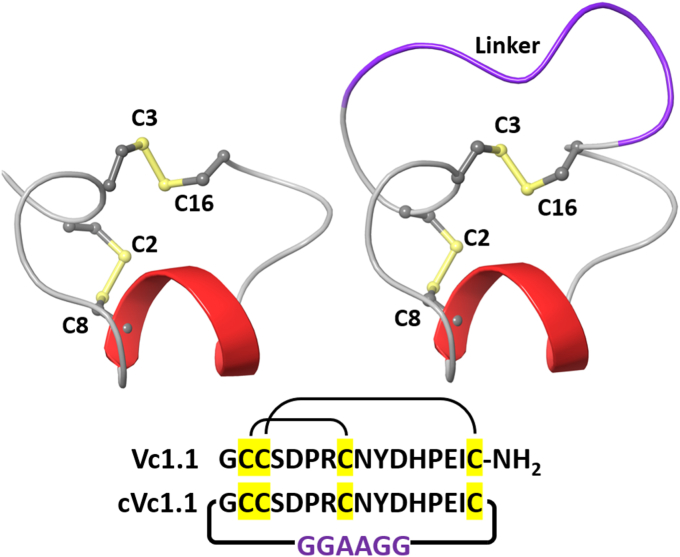
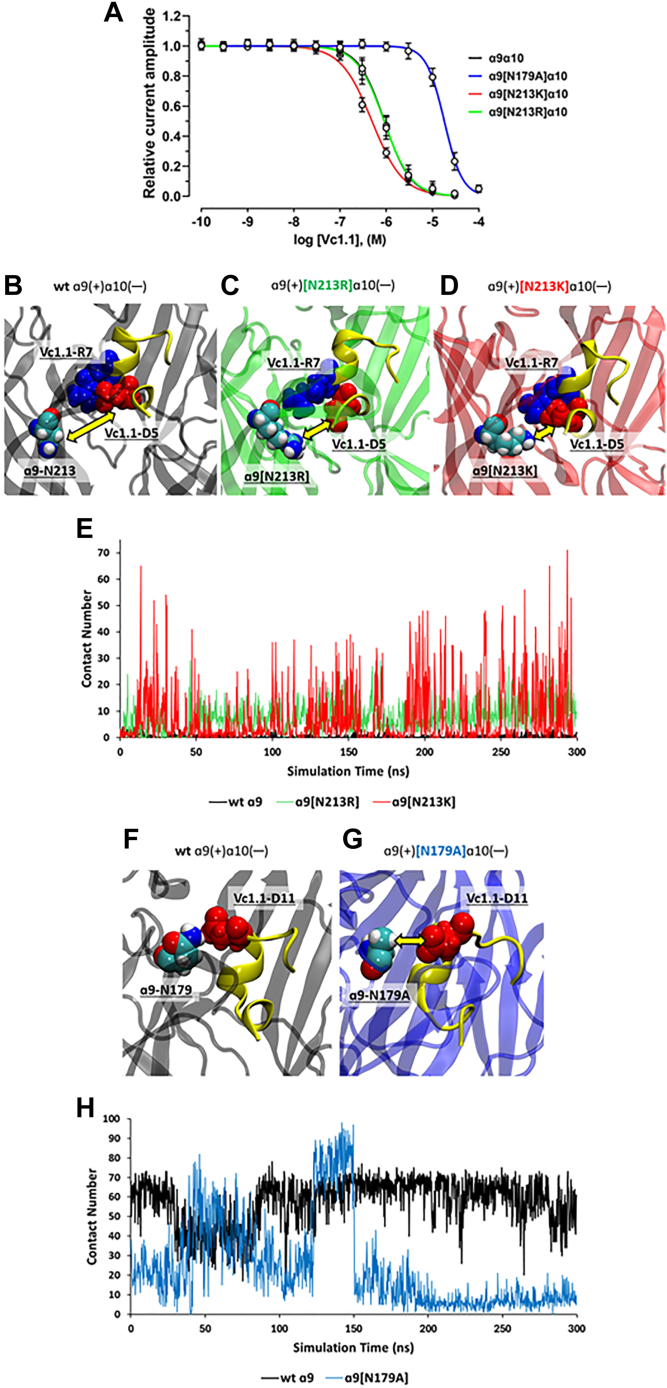
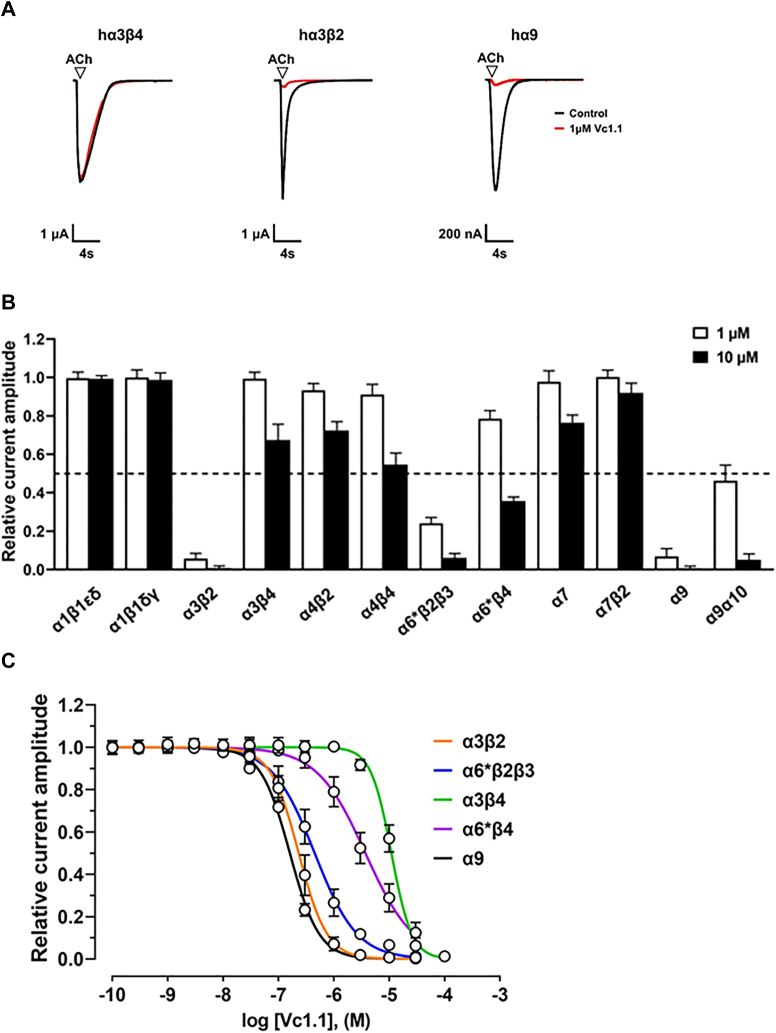
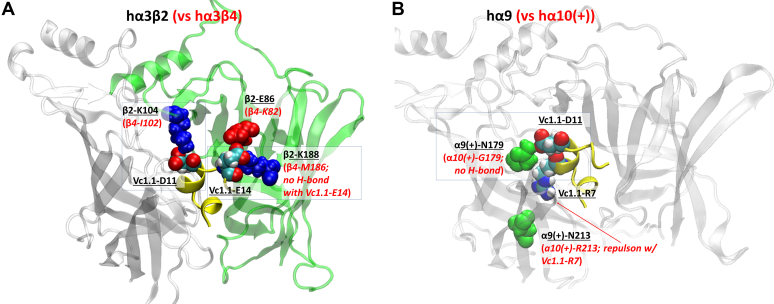
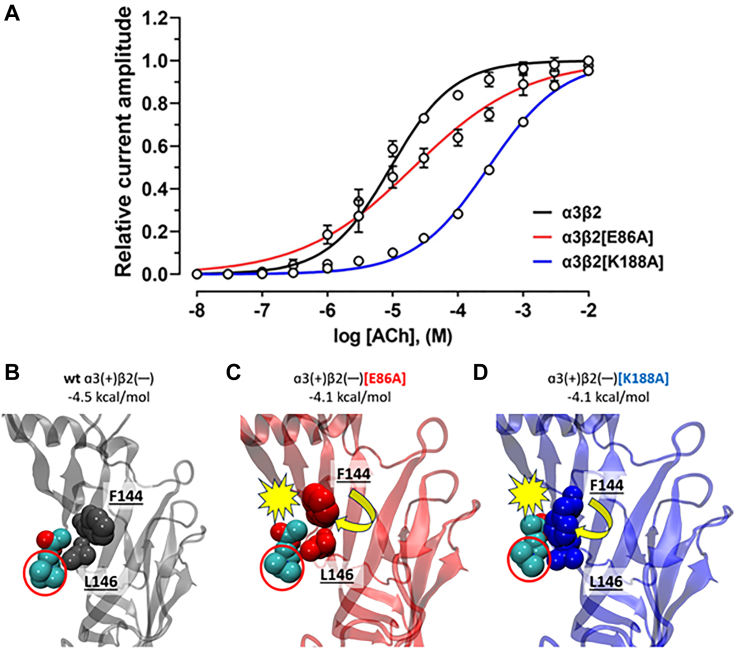
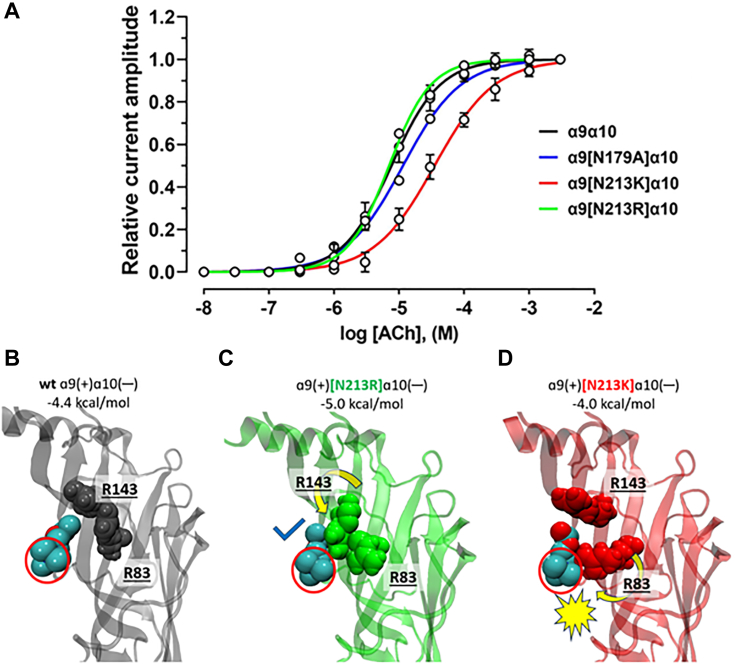
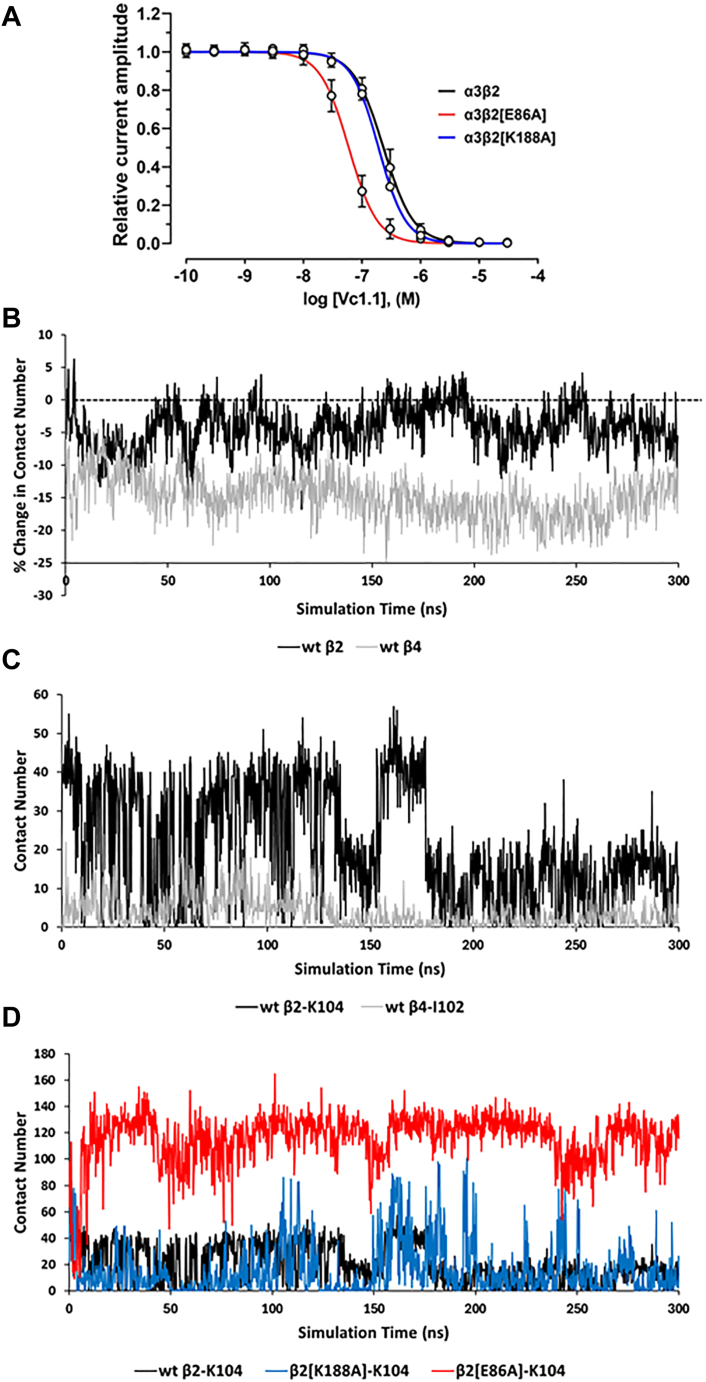
The α-conotoxins (α-Ctxs) are short, disulfide-rich peptides derived from the venom of the Conus marine snails, primarily acting as antagonists of nicotinic acetylcholine receptors (nAChRs). Specifically, α-Ctx Vc1.1, a 16-amino acid peptide from Conus victoriae, competitively antagonizes non-muscle nAChRs, inhibits nicotine-induced currents in bovine chromaffin cells, and alleviates neuropathic pain in rat models. Although Vc1.1 selectively inhibits rat α9α10 nAChRs, its potency and selectivity across human nAChR subtypes remain unresolved. In this study, we assessed the activity of Vc1.1 on human (h) nAChRs heterologously expressed in Xenopus laevis oocytes using the two-electrode voltage clamp technique and simulated interactions using computational modeling. Vc1.1 selectively antagonized homomeric α9 and heteromeric α3β2 nAChRs, with half-maximal inhibitory concentrations (IC) of 160 nM and 232 nM, respectively. At hα9[N179A]α10, Vc1.1 exhibited a 20-fold decrease in potency compared to hα9α10, due to the loss of hydrogen bonding with Vc1.1-D11. Conversely, Vc1.1 was four-fold more potent at hα3β2[E86A] compared to hα3β2, possibly influenced by the proximal residue β2-K104, as suggested by molecular dynamics (MD) simulations. Additionally, Vc1.1's potency doubled at hα9[N213K]α10, whereas it remained unchanged at hα9[N213R]α10 nAChRs. MD simulations indicate that altered interactions between the mutant hα9 N179A, N213K, and N213R side chains and Vc1.1-D5 may partly explain these changes in potency. The inhibitory action of Vc1.1 at α9-containing nAChRs is particularly relevant given their role in neuroinflammation, presenting a potential therapeutic pathway for alleviating neuropathic and inflammatory pain. This study provides valuable insights into the rational design of Vc1.1-derived α-Ctxs with enhanced nAChR subtype selectivity.
α-芋螺毒素(α-Ctxs)是源自芋螺属海洋蜗牛毒液的短链、富含二硫键的肽,主要作为烟碱型乙酰胆碱受体(nAChRs)的拮抗剂。具体而言,来自维多利亚芋螺的16个氨基酸的肽α-Ctx Vc1.1竞争性拮抗非肌肉型nAChRs,抑制牛嗜铬细胞中尼古丁诱导的电流,并减轻大鼠模型中的神经性疼痛。尽管Vc1.1选择性抑制大鼠α9α10 nAChRs,但其对人nAChR亚型的效力和选择性仍未明确。在本研究中,我们使用双电极电压钳技术评估了Vc1.1对非洲爪蟾卵母细胞中异源表达的人(h)nAChRs的活性,并使用计算模型模拟了相互作用。Vc1.1选择性拮抗同源性α9和异源性α3β2 nAChRs,半数最大抑制浓度(IC)分别为160 nM和232 nM。在hα9[N179A]α10上,与hα9α10相比,Vc1.1的效力降低了20倍,这是由于与Vc1.1-D11的氢键丢失所致。相反,与hα3β2相比,Vc1.1在hα3β2[E86A]上的效力高4倍,分子动力学(MD)模拟表明,这可能受近端残基β2-K104的影响。此外,Vc1.1在hα9[N213K]α10上的效力加倍,而在hα9[N213R]α10 nAChRs上保持不变。MD模拟表明,突变型hα9 N179A、N213K和N213R侧链与Vc1.1-D5之间相互作用的改变可能部分解释了这些效力变化。鉴于其在神经炎症中的作用,Vc1.1对含α9的nAChRs的抑制作用尤为重要,为减轻神经性和炎症性疼痛提供了一条潜在的治疗途径。本研究为合理设计具有增强的nAChR亚型选择性的Vc1.1衍生的α-Ctxs提供了有价值的见解。