Zhu Li-Fang, Körmann Fritz, Chen Qing, Selleby Malin, Neugebauer Jörg, Grabowski Blazej
Department for Computational Materials Design, Max Planck Institute for Sustainable Materials, Max-Planck-str.1, 40237 Düsseldorf, Germany.
Institute for Materials Science, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany.
NPJ Comput Mater. 2024;10(1):274. doi: 10.1038/s41524-024-01464-7. Epub 2024 Nov 29.
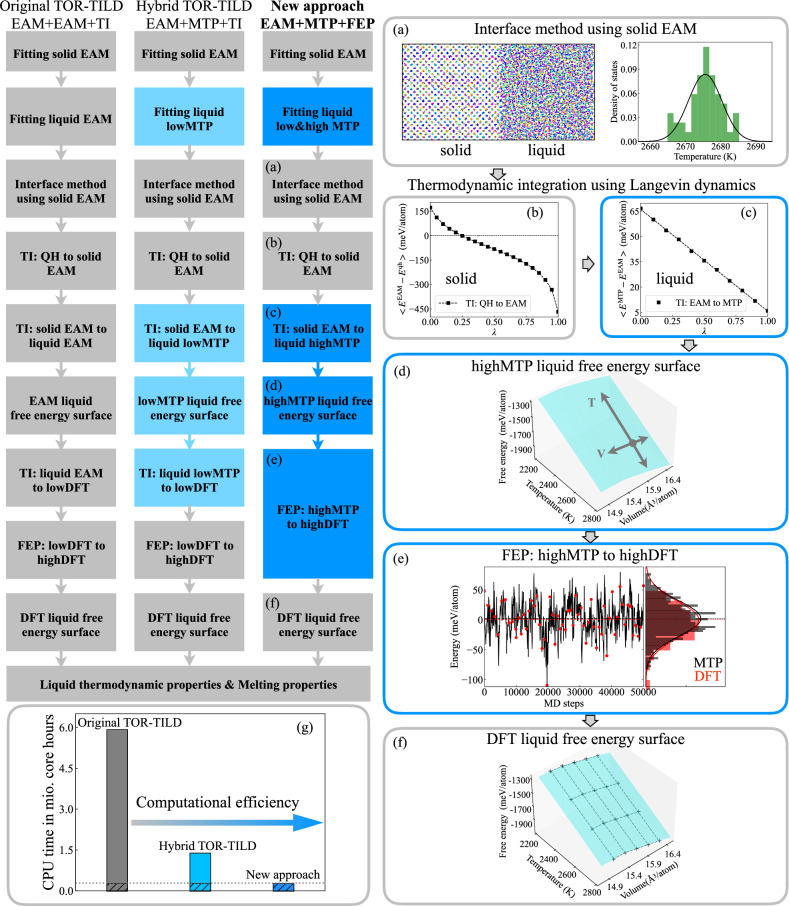
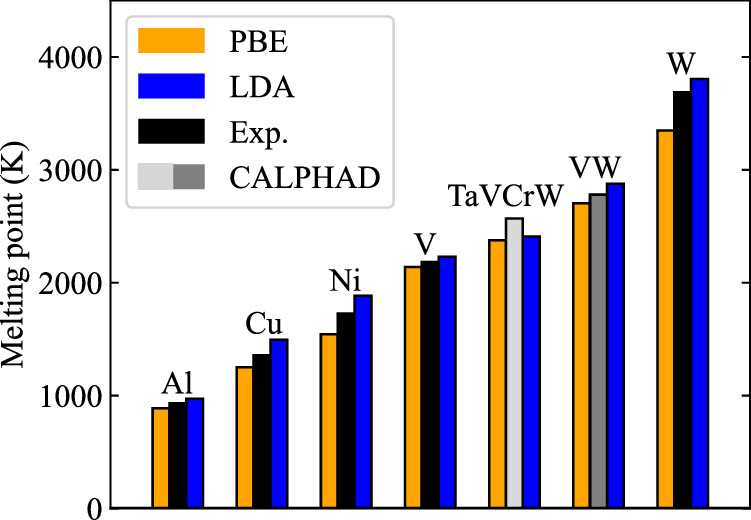
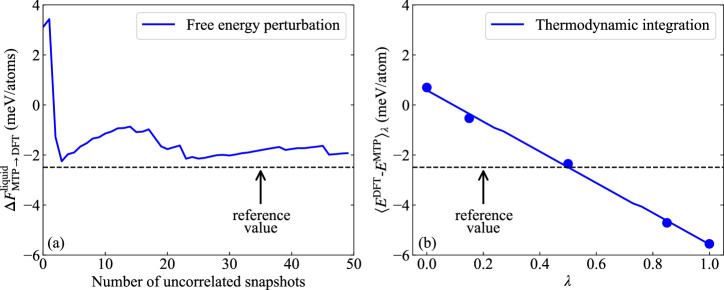
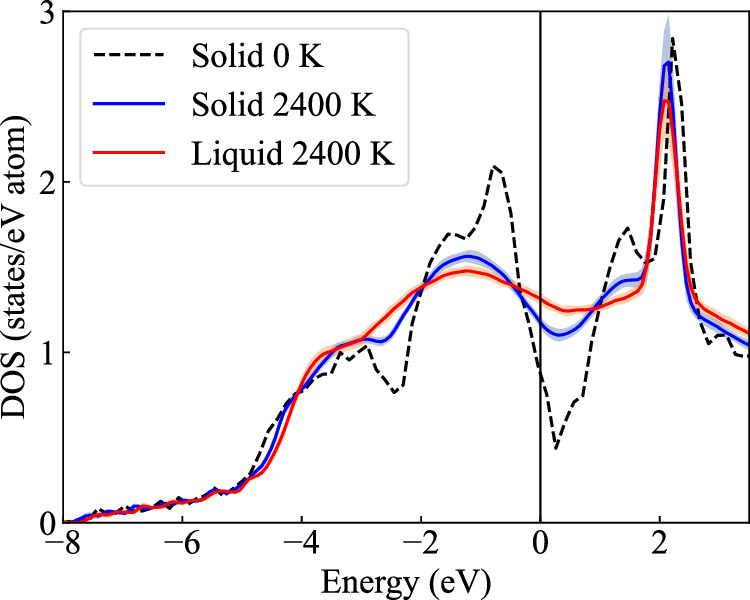
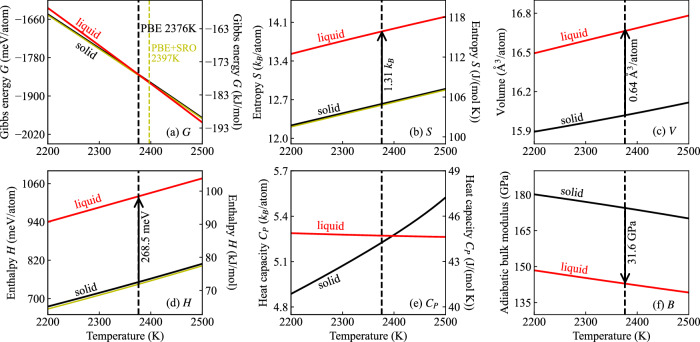
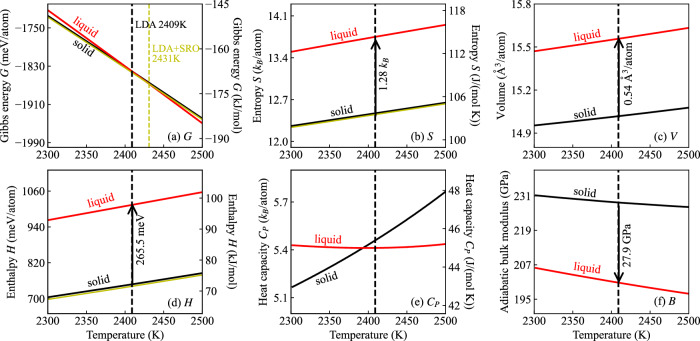
Melting properties are critical for designing novel materials, especially for discovering high-performance, high-melting refractory materials. Experimental measurements of these properties are extremely challenging due to their high melting temperatures. Complementary theoretical predictions are, therefore, indispensable. One of the most accurate approaches for this purpose is the ab initio free-energy approach based on density functional theory (DFT). However, it generally involves expensive thermodynamic integration using ab initio molecular dynamic simulations. The high computational cost makes high-throughput calculations infeasible. Here, we propose a highly efficient DFT-based method aided by a specially designed machine learning potential. As the machine learning potential can closely reproduce the ab initio phase-space distribution, even for multi-component alloys, the costly thermodynamic integration can be fully substituted with more efficient free energy perturbation calculations. The method achieves overall savings of computational resources by 80% compared to current alternatives. We apply the method to the high-entropy alloy TaVCrW and calculate its melting properties, including the melting temperature, entropy and enthalpy of fusion, and volume change at the melting point. Additionally, the heat capacities of solid and liquid TaVCrW are calculated. The results agree reasonably with the CALPHAD extrapolated values.
熔化特性对于新型材料的设计至关重要,特别是对于发现高性能、高熔点的难熔材料而言。由于这些材料的熔点很高,对其特性进行实验测量极具挑战性。因此,互补的理论预测必不可少。为此,最精确的方法之一是基于密度泛函理论(DFT)的从头算自由能方法。然而,它通常涉及使用从头算分子动力学模拟进行昂贵的热力学积分。高计算成本使得高通量计算变得不可行。在此,我们提出一种基于DFT的高效方法,借助专门设计的机器学习势。由于机器学习势能够紧密再现从头算相空间分布,即使对于多组分合金也是如此,因此可以用更高效的自由能微扰计算完全替代昂贵的热力学积分。与现有方法相比,该方法总体节省了80%的计算资源。我们将该方法应用于高熵合金TaVCrW,并计算其熔化特性,包括熔化温度、熔化熵和焓以及熔点处的体积变化。此外,还计算了固态和液态TaVCrW的热容。结果与CALPHAD外推值合理相符。