Hwang Taejoo, Sitko Lukasz Karol, Khoirunnisa Ratih, Navarro-Aguad Fernanda, Samuel David M, Park Hajoong, Cheon Banyoon, Mutsnaini Luthfiyyah, Lee Jaewoong, Otlu Burçak, Takeda Shunichi, Lee Semin, Ivanov Dmitri, Gartner Anton
Department of Biomedical Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan, 44919, Republic of Korea.
Department of Biological Sciences, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 44919, Republic of Korea.
Nucleic Acids Res. 2025 Jan 7;53(1). doi: 10.1093/nar/gkae1122.
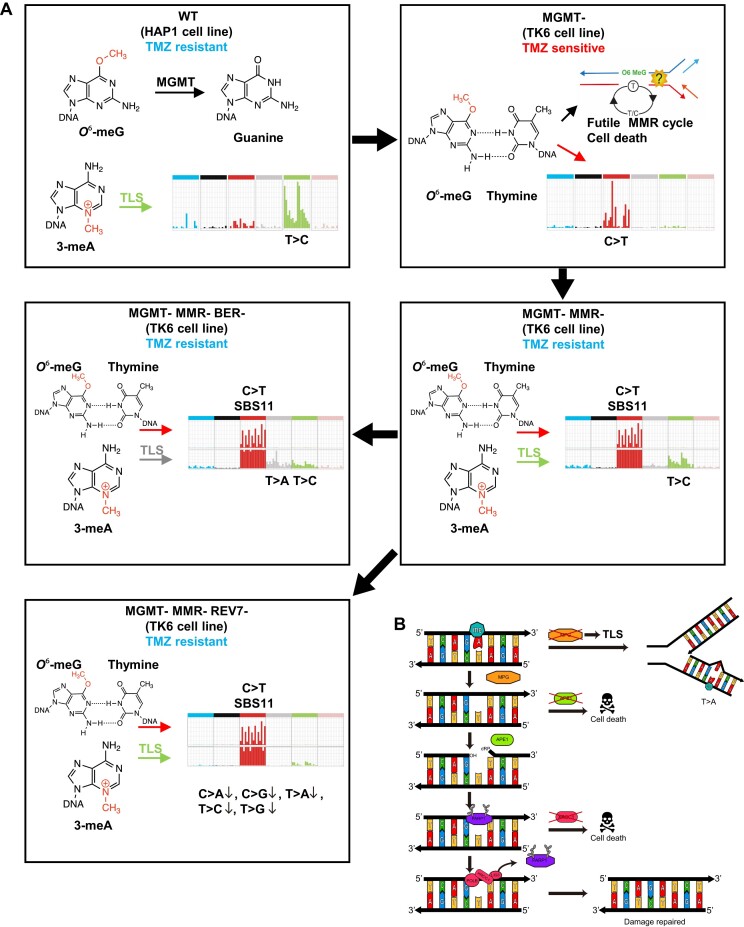
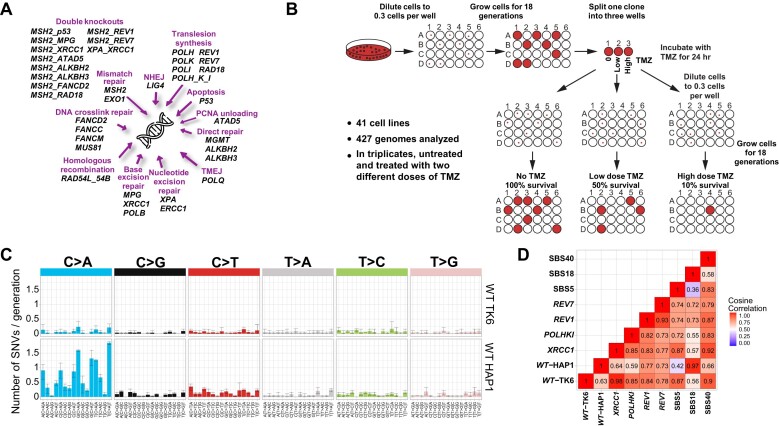
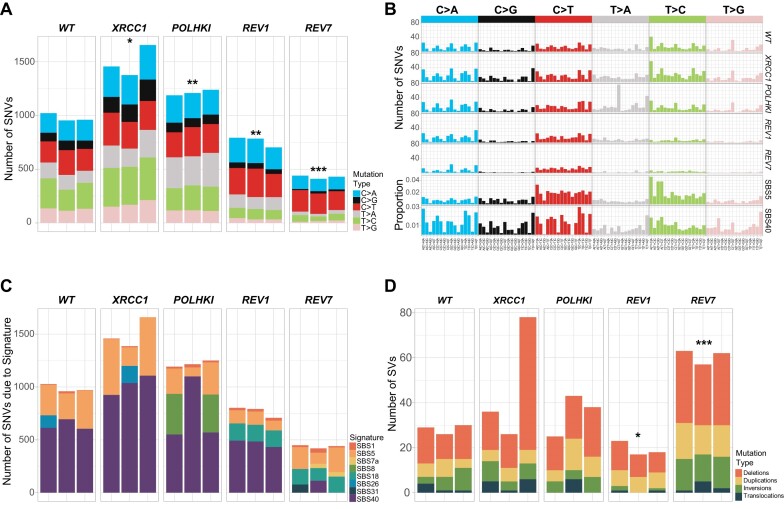
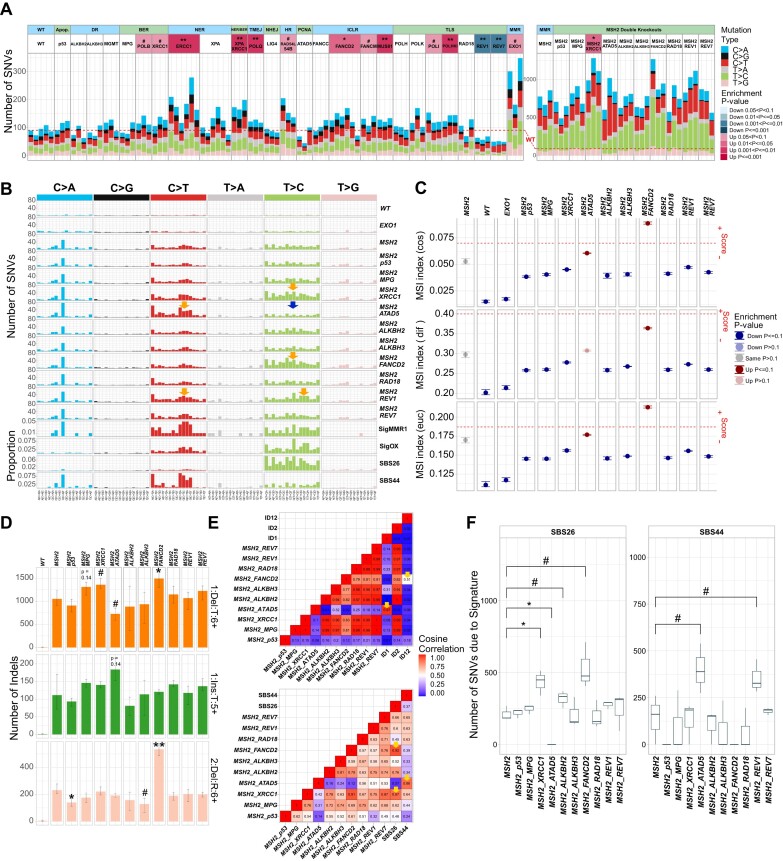
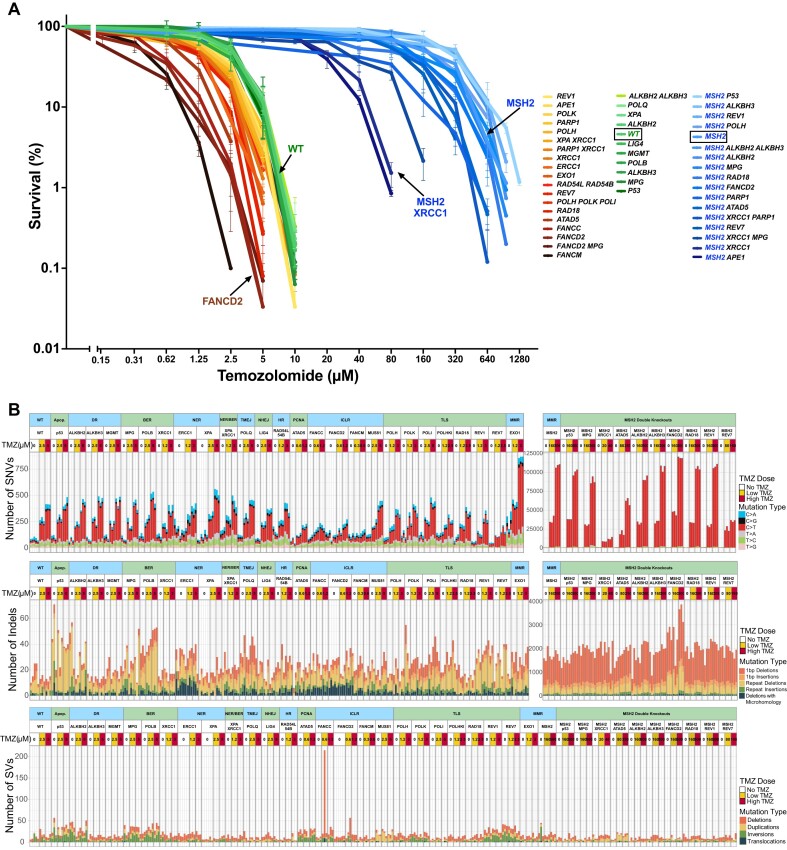
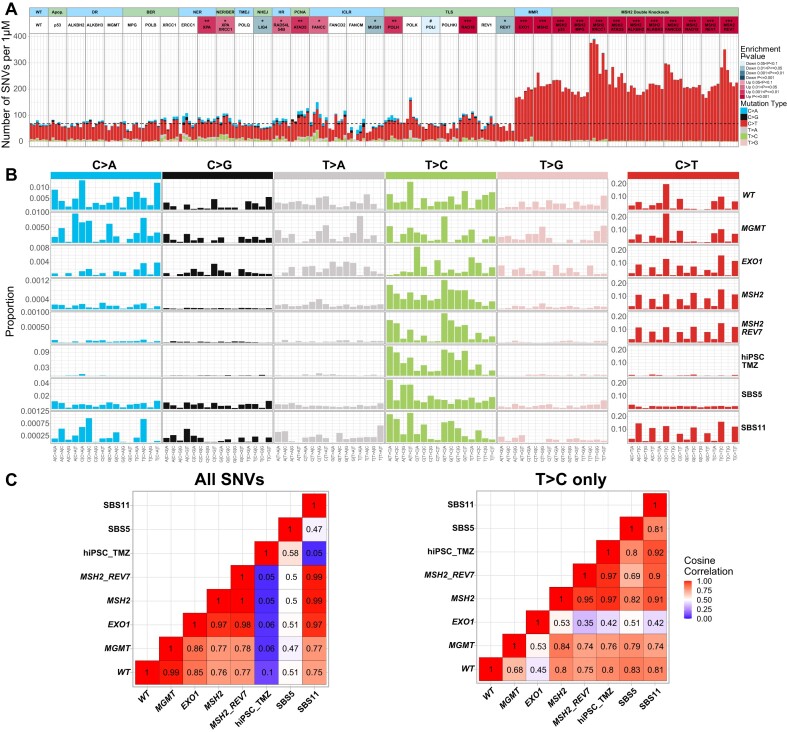
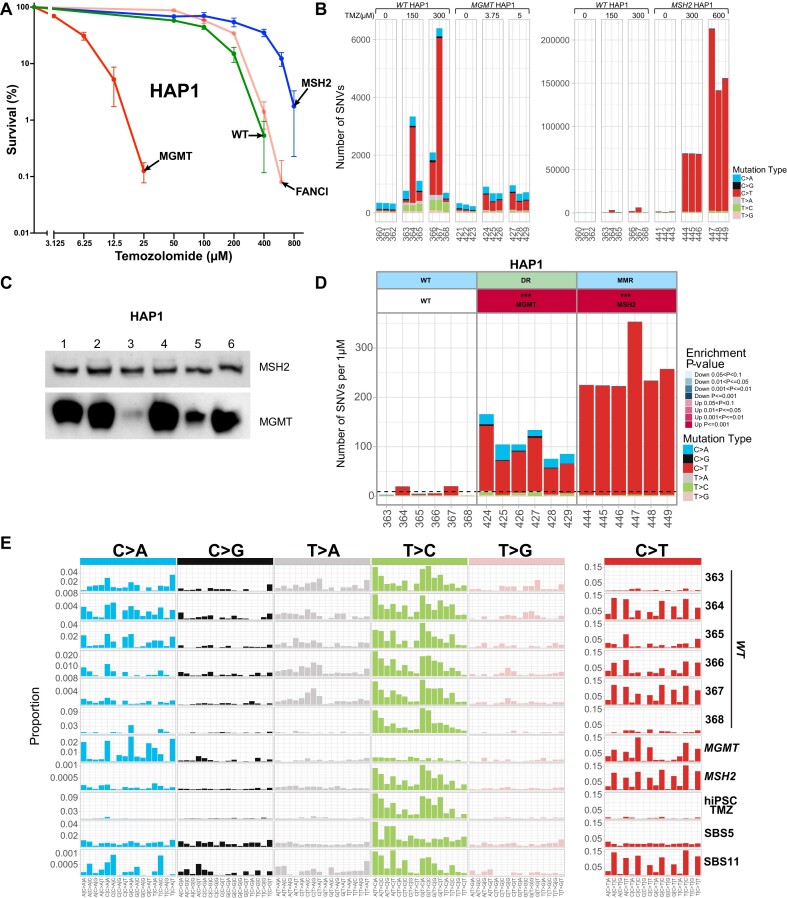
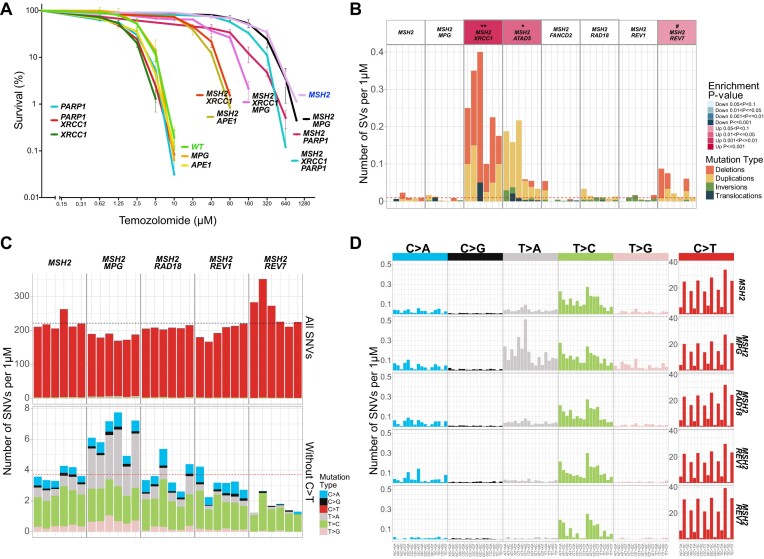
In a comprehensive study to decipher the multi-layered response to the chemotherapeutic agent temozolomide (TMZ), we analyzed 427 genomes and determined mutational patterns in a collection of ∼40 isogenic DNA repair-deficient human TK6 lymphoblast cell lines. We first demonstrate that the spontaneous mutational background is very similar to the aging-associated mutational signature SBS40 and mainly caused by polymerase zeta-mediated translesion synthesis (TLS). MSH2-/- mismatch repair (MMR) knockout in conjunction with additional repair deficiencies uncovers cryptic mutational patterns. We next report how distinct mutational signatures are induced by TMZ upon sequential inactivation of DNA repair pathways, mirroring the acquisition of chemotherapy resistance by glioblastomas. The most toxic adduct induced by TMZ, O6-meG, is directly repaired by the O6-methylguanine-DNA methyltransferase (MGMT). In MGMT-/- cells, MMR leads to cell death and limits mutagenesis. MMR deficiency results in TMZ resistance, allowing the accumulation of ∼105 C > T substitutions corresponding to signature SBS11. Under these conditions, N3-methyladenine (3-meA), processed by base excision repair (BER), limits cell survival. Without BER, 3-meA is read through via error-prone TLS, causing T > A substitutions but not affecting survival. Blocking BER after abasic site formation results in large deletions and TMZ hypersensitization. Our findings reveal potential vulnerabilities of TMZ-resistant tumors.
在一项旨在解析对化疗药物替莫唑胺(TMZ)的多层次反应的综合研究中,我们分析了427个基因组,并确定了约40个同基因DNA修复缺陷型人TK6淋巴母细胞系集合中的突变模式。我们首先证明,自发突变背景与衰老相关的突变特征SBS40非常相似,主要由聚合酶ζ介导的跨损伤合成(TLS)引起。MSH2基因敲除导致错配修复(MMR)缺陷,并伴有其他修复缺陷,从而揭示了隐秘的突变模式。接下来,我们报告了在DNA修复途径相继失活后,TMZ如何诱导不同的突变特征,这反映了胶质母细胞瘤获得化疗耐药性的过程。TMZ诱导的最具毒性的加合物O6-甲基鸟嘌呤(O6-meG)由O6-甲基鸟嘌呤-DNA甲基转移酶(MGMT)直接修复。在MGMT缺陷型细胞中,MMR会导致细胞死亡并限制诱变。MMR缺陷导致TMZ耐药,使得对应于特征SBS11的约105个C>T替换得以积累。在这些条件下,由碱基切除修复(BER)处理的N3-甲基腺嘌呤(3-meA)会限制细胞存活。没有BER时,3-meA会通过易出错的TLS通读,导致T>A替换,但不影响细胞存活。在无碱基位点形成后阻断BER会导致大片段缺失和TMZ超敏反应。我们的研究结果揭示了TMZ耐药肿瘤潜在的脆弱性。