Ahn Sangwook, Redman Elizabeth M, Gavriliuc Stefan, Bellaw Jennifer, Gilleard John S, McLoughlin Philip D, Poissant Jocelyn
Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada.
M.H. Gluck Equine Research Center, Department of Veterinary Science, University of Kentucky, Lexington, KY, USA.
Parasitology. 2024 Oct;151(12):1299-1316. doi: 10.1017/S0031182024001185. Epub 2024 Dec 12.
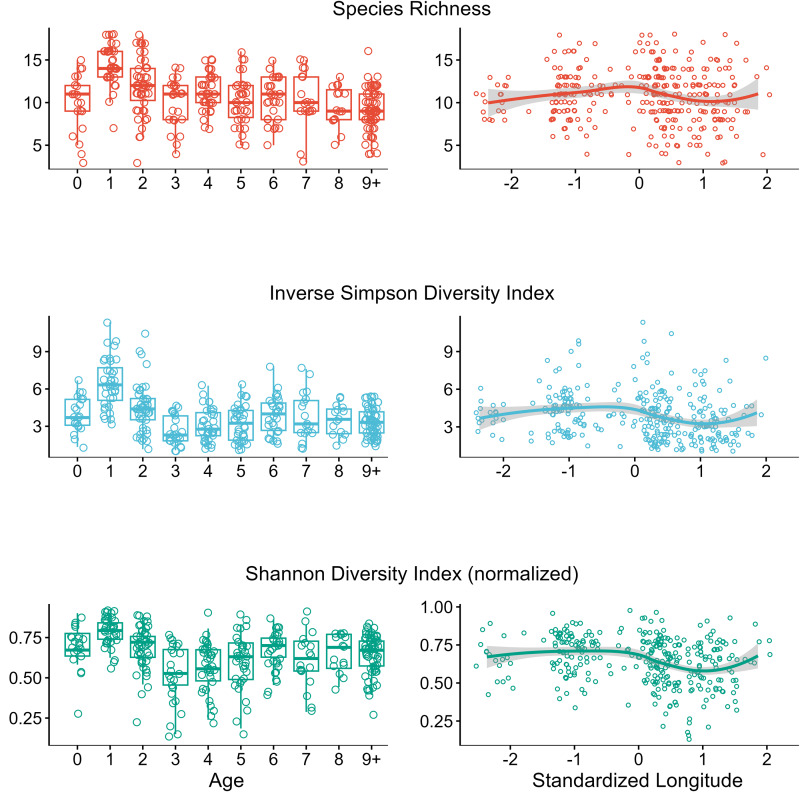
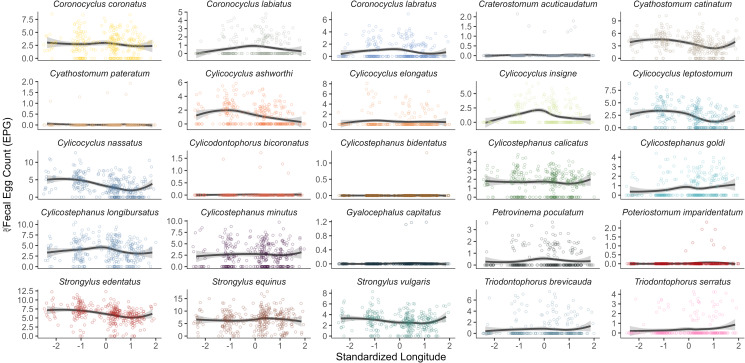
Identifying factors that drive among-individual variation in mixed parasitic infections is fundamental to understanding the ecology and evolution of host–parasite interactions. However, a lack of non-invasive diagnostic tools to quantify mixed infections has restricted their investigation for host populations in the wild. This study applied DNA metabarcoding on parasite larvae cultured from faecal samples to characterize mixed strongyle infections of 320 feral horses on Sable Island, Nova Scotia, Canada, in 2014 to test for the influence of host (age, sex and reproductive/social status) and environmental (location, local density and social group membership) factors on variation. Twenty-five strongyle species were identified, with individual infections ranging from 3 to 18 species with a mean richness (±1 s.d.) of 10.8 ± 3.1. Strongyle eggs shed in faeces were dominated by small strongyle (cyathostomins) species in young individuals, transitioning to large strongyles ( spp.) in adults. Egg counts were highest in young individuals and in the west or centre of the island for most species. Individuals in the same social group had similar parasite communities, supporting the hypothesis that shared environment may drive parasite assemblages. Other factors such as local horse density, sex, date and reproductive/social status had minimal impacts on infection patterns. This study demonstrates that mixed infections can be dynamic across host ontogeny and space and emphasizes the need to consider species-specific infection patterns when investigating mixed infections.
确定导致混合寄生虫感染个体间差异的因素,对于理解宿主 - 寄生虫相互作用的生态学和进化至关重要。然而,缺乏用于量化混合感染的非侵入性诊断工具,限制了对野生宿主种群的此类研究。本研究对从粪便样本中培养出的寄生虫幼虫进行DNA宏条形码分析,以表征2014年加拿大新斯科舍省黑貂岛上320匹野马的混合圆线虫感染情况,测试宿主(年龄、性别和生殖/社会地位)和环境(位置、当地密度和社会群体成员身份)因素对差异的影响。共鉴定出25种圆线虫,个体感染的种类从3种到18种不等,平均丰富度(±1标准差)为10.8±3.1。粪便中排出的圆线虫卵在幼体中以小型圆线虫(杯口线虫)为主,成年后转变为大型圆线虫(多种)。大多数种类的虫卵计数在幼体以及岛屿西部或中部最高。同一社会群体中的个体具有相似的寄生虫群落,支持了共享环境可能驱动寄生虫组合的假设。其他因素,如当地马匹密度、性别、日期和生殖/社会地位,对感染模式的影响最小。本研究表明,混合感染在宿主个体发育和空间上可能是动态变化的,并强调在研究混合感染时需要考虑物种特异性感染模式。