Horvath Jakub, Jedlicka Pavel, Kratka Marie, Kubat Zdenek, Kejnovsky Eduard, Lexa Matej
Faculty of Informatics, Masaryk University, Botanicka 68a, Brno, 60200, Czech Republic.
Department of Plant Developmental Genetics, Institute of Biophysics of the Czech Academy of Sciences, Kralovopolska 135, Brno, 61200, Czech Republic.
BioData Min. 2024 Dec 18;17(1):57. doi: 10.1186/s13040-024-00410-z.
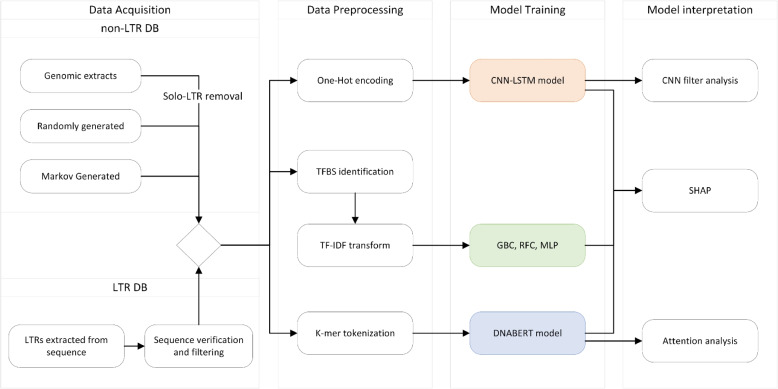
Long terminal repeats (LTRs) represent important parts of LTR retrotransposons and retroviruses found in high copy numbers in a majority of eukaryotic genomes. LTRs contain regulatory sequences essential for the life cycle of the retrotransposon. Previous experimental and sequence studies have provided only limited information about LTR structure and composition, mostly from model systems. To enhance our understanding of these key sequence modules, we focused on the contrasts between LTRs of various retrotransposon families and other genomic regions. Furthermore, this approach can be utilized for the classification and prediction of LTRs.
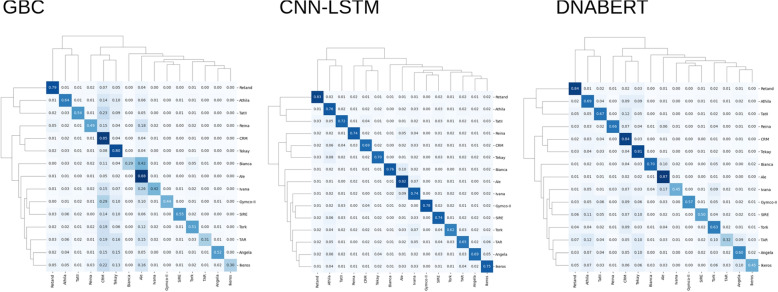
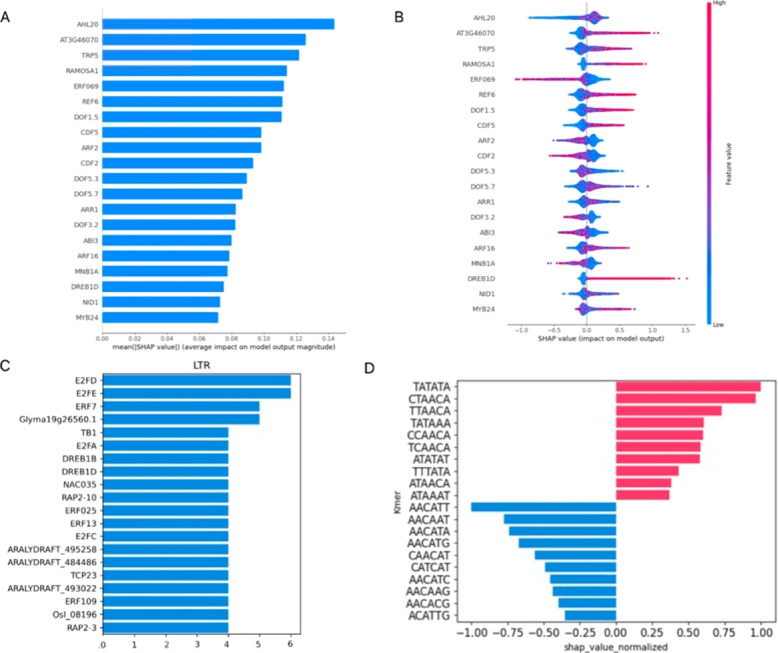
We used machine learning methods suitable for DNA sequence classification and applied them to a large dataset of plant LTR retrotransposon sequences. We trained three machine learning models using (i) traditional model ensembles (Gradient Boosting), (ii) hybrid convolutional/long and short memory network models, and (iii) a DNA pre-trained transformer-based model using k-mer sequence representation. All three approaches were successful in classifying and isolating LTRs in this data, as well as providing valuable insights into LTR sequence composition. The best classification (expressed as F1 score) achieved for LTR detection was 0.85 using the hybrid network model. The most accurate classification task was superfamily classification (F1=0.89) while the least accurate was family classification (F1=0.74). The trained models were subjected to explainability analysis. Positional analysis identified a mixture of interesting features, many of which had a preferred absolute position within the LTR and/or were biologically relevant, such as a centrally positioned TATA-box regulatory sequence, and TG..CA nucleotide patterns around both LTR edges.
Our results show that the models used here recognized biologically relevant motifs, such as core promoter elements in the LTR detection task, and a development and stress-related subclass of transcription factor binding sites in the family classification task. Explainability analysis also highlighted the importance of 5'- and 3'- edges in LTR identity and revealed need to analyze more than just dinucleotides at these ends. Our work shows the applicability of machine learning models to regulatory sequence analysis and classification, and demonstrates the important role of the identified motifs in LTR detection.
长末端重复序列(LTRs)是LTR逆转录转座子和逆转录病毒的重要组成部分,在大多数真核生物基因组中以高拷贝数存在。LTRs包含逆转录转座子生命周期所必需的调控序列。先前的实验和序列研究仅提供了关于LTR结构和组成的有限信息,大多来自模型系统。为了加深我们对这些关键序列模块的理解,我们重点研究了各种逆转录转座子家族的LTRs与其他基因组区域之间的差异。此外,这种方法可用于LTRs的分类和预测。
我们使用了适用于DNA序列分类的机器学习方法,并将其应用于植物LTR逆转录转座子序列的大型数据集。我们使用(i)传统模型集成(梯度提升)、(ii)混合卷积/长短时记忆网络模型和(iii)使用k-mer序列表示的基于DNA预训练的Transformer模型训练了三种机器学习模型。这三种方法在对该数据中的LTRs进行分类和分离方面均取得成功,并为LTR序列组成提供了有价值的见解。使用混合网络模型进行LTR检测时,获得的最佳分类(以F1分数表示)为0.85。最准确的分类任务是超家族分类(F1 = 0.89),而最不准确的是家族分类(F1 = 0.74)。对训练好的模型进行了可解释性分析。位置分析确定了一系列有趣的特征,其中许多在LTR内具有优先的绝对位置和/或具有生物学相关性,例如位于中心位置的TATA框调控序列,以及LTR两侧边缘的TG..CA核苷酸模式。
我们的结果表明,这里使用的模型识别出了生物学相关的基序,例如LTR检测任务中的核心启动子元件,以及家族分类任务中与发育和应激相关的转录因子结合位点亚类。可解释性分析还突出了5'和3'边缘在LTR识别中的重要性,并揭示了不仅需要分析这些末端的二核苷酸。我们的工作展示了机器学习模型在调控序列分析和分类中的适用性,并证明了所识别基序在LTR检测中的重要作用。