Zhao Jianshu, Pierre Both Jean, Konstantinidis Konstantinos T
Center for Bioinformatics and Computational Genomics, Georgia Institute of Technology, 225 North Avenue NW, Atlanta, GA, 30332, USA.
School of Biological Sciences, Georgia Institute of Technology, 225 North Avenue NW, Atlanta, GA, 30332, USA.
NAR Genom Bioinform. 2024 Dec 18;6(4):lqae172. doi: 10.1093/nargab/lqae172. eCollection 2024 Dec.
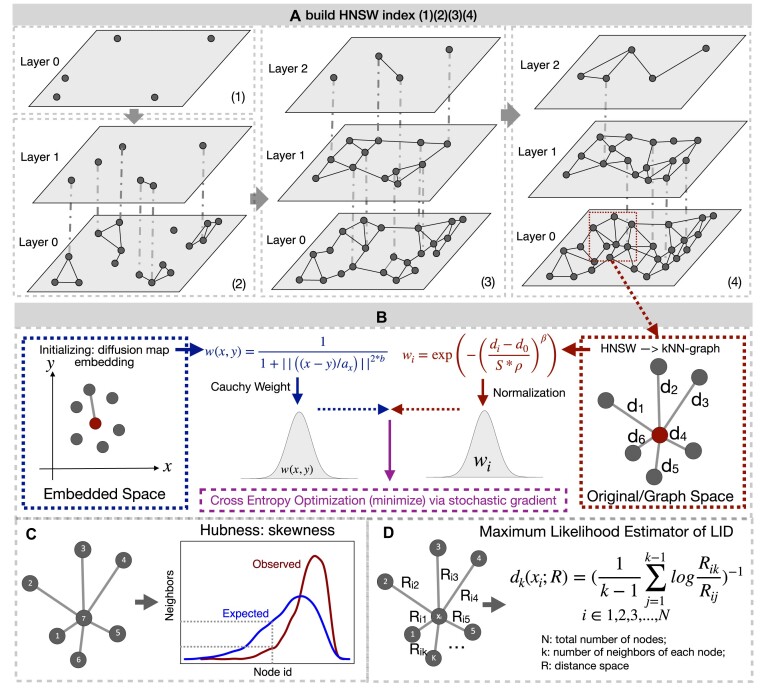
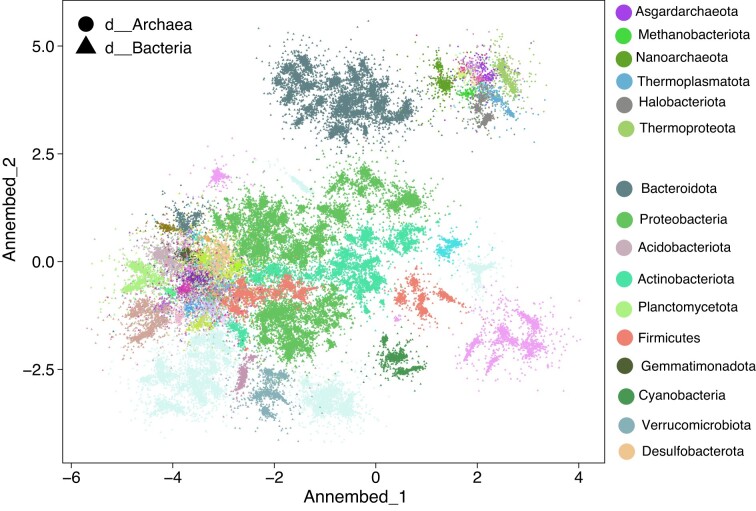
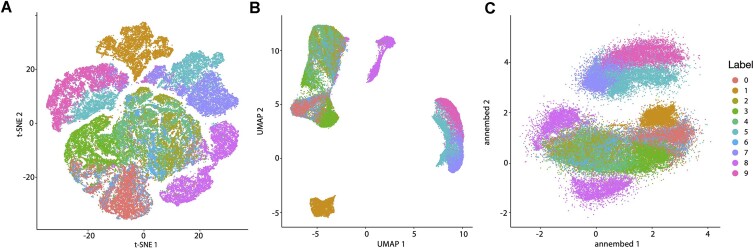
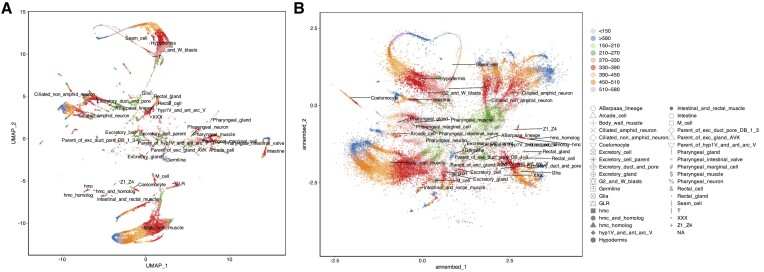
Dimension reduction (DR or embedding) algorithms such as t-SNE and UMAP have many applications in big data visualization but remain slow for large datasets. Here, we further improve the UMAP-like algorithms by (i) combining several aspects of t-SNE and UMAP to create a new DR algorithm; (ii) replacing its rate-limiting step, the K-nearest neighbor graph (K-NNG), with a Hierarchical Navigable Small World (HNSW) graph; and (iii) extending the functionality to DNA/RNA sequence data by combining HNSW with locality sensitive hashing algorithms (e.g. MinHash) for distance estimations among sequences. We also provide additional features including computation of local intrinsic dimension and hubness, which can reflect structures and properties of the underlying data that strongly affect the K-NNG accuracy, and thus the quality of the resulting embeddings. Our library, called annembed, is implemented, and fully parallelized in Rust and shows competitive accuracy compared to the popular UMAP-like algorithms. Additionally, we showcase the usefulness and scalability of our library with three real-world examples: visualizing a large-scale microbial genomic database, visualizing single-cell RNA sequencing data and metagenomic contig (or population) binning. Therefore, annembed can facilitate DR for several tasks for biological data analysis where distance computation is expensive or when there are millions to billions of data points to process.
诸如t-SNE和UMAP之类的降维(DR或嵌入)算法在大数据可视化中有许多应用,但对于大型数据集而言仍然速度较慢。在此,我们通过以下方式进一步改进类UMAP算法:(i)结合t-SNE和UMAP的多个方面来创建一种新的DR算法;(ii)用分层可导航小世界(HNSW)图替换其限速步骤——K近邻图(K-NNG);(iii)通过将HNSW与局部敏感哈希算法(如MinHash)相结合以进行序列间距离估计,从而将功能扩展到DNA/RNA序列数据。我们还提供了其他功能,包括局部固有维度和中心性的计算,这些可以反映强烈影响K-NNG准确性进而影响所得嵌入质量的基础数据的结构和属性。我们名为annembed的库已用Rust实现并完全并行化,与流行的类UMAP算法相比显示出具有竞争力的准确性。此外,我们通过三个实际示例展示了我们库的实用性和可扩展性:可视化大规模微生物基因组数据库、可视化单细胞RNA测序数据以及宏基因组重叠群(或群体)分箱。因此,annembed可以促进生物数据分析中若干任务的降维,在这些任务中距离计算成本高昂,或者存在数百万到数十亿个数据点需要处理。