Wang Huarui, Sun Chuqing, Li Yun, Chen Jingchao, Zhao Xing-Ming, Chen Wei-Hua
Key Laboratory of Molecular Biophysics of the Ministry of Education, Hubei Key Laboratory of Bioinformatics and Molecular Imaging, Department of Bioinformatics and Systems Biology, Center for Artificial Intelligence Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 430074, China.
Department of Neurology, Institute of Science and Technology for Brain-Inspired Intelligence, Zhongshan Hospitaland, Fudan University , Shanghai, 200433, China.
Microbiome. 2024 Dec 20;12(1):260. doi: 10.1186/s40168-024-01981-z.
Metagenome-assembled viral genomes have significantly advanced the discovery and characterization of the human gut virome. However, we lack a comparative assessment of assembly tools on the efficacy of viral genome identification, particularly across next-generation sequencing (NGS) and third-generation sequencing (TGS) data.
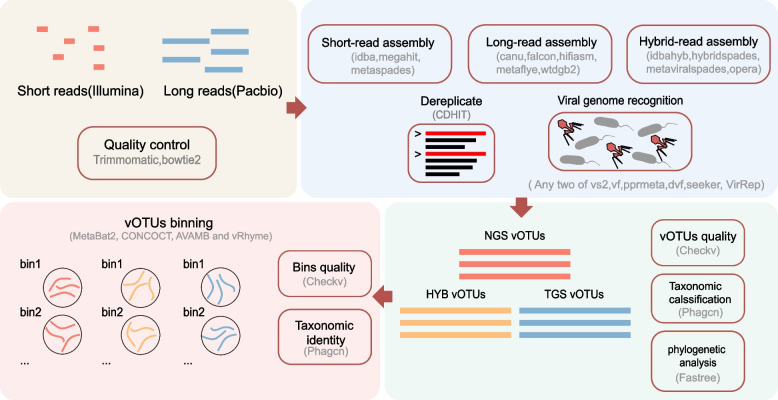
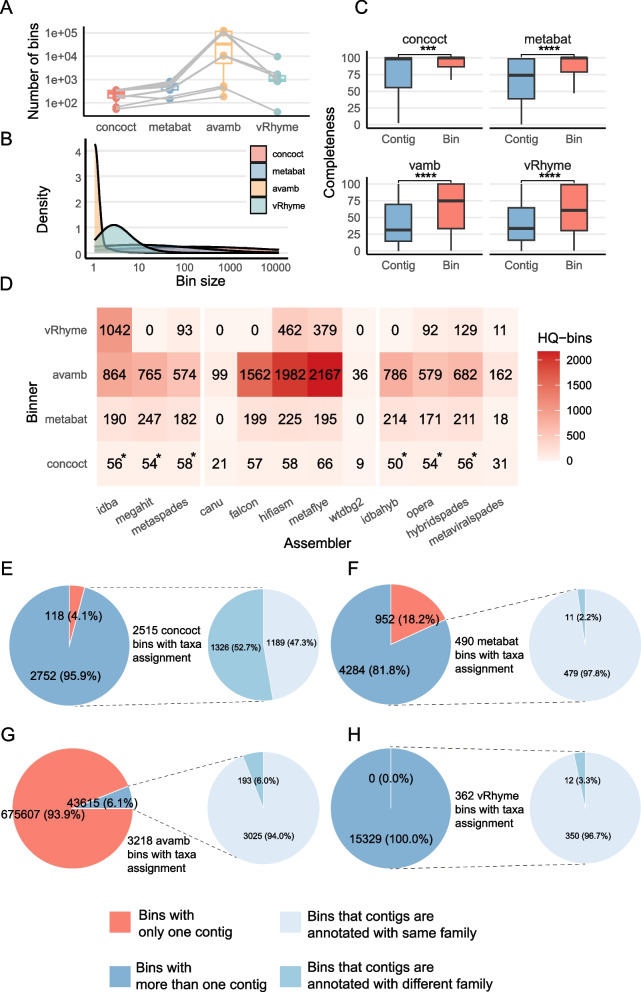
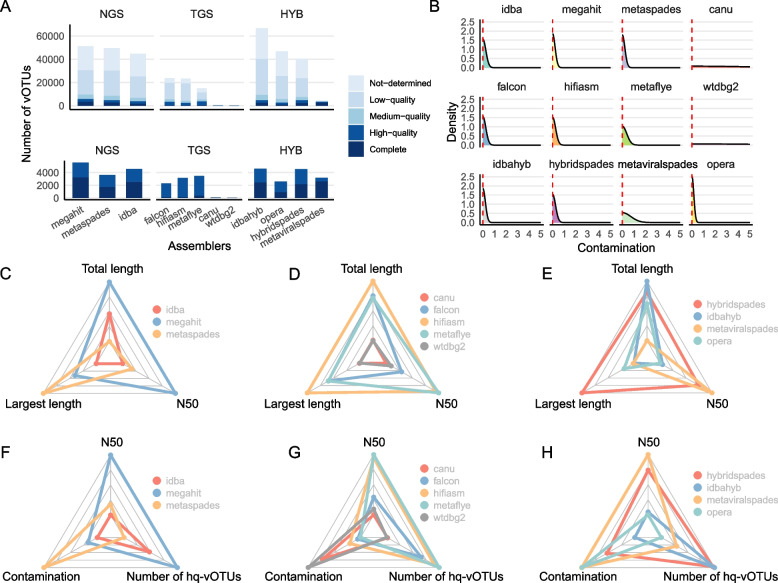
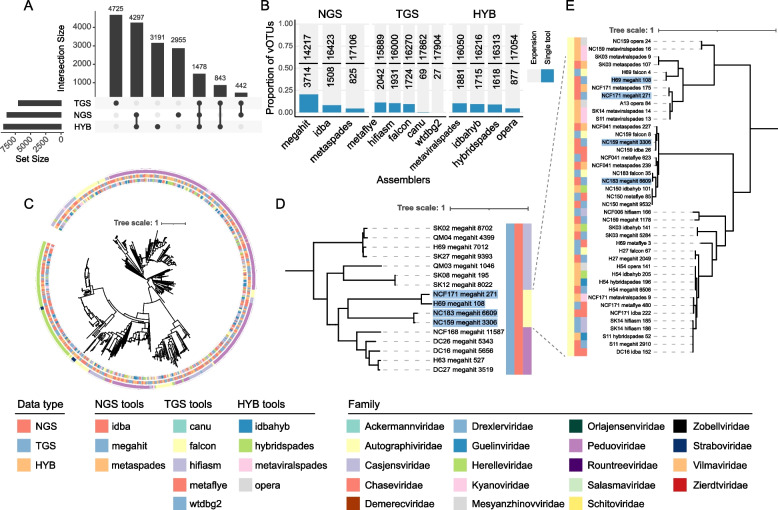
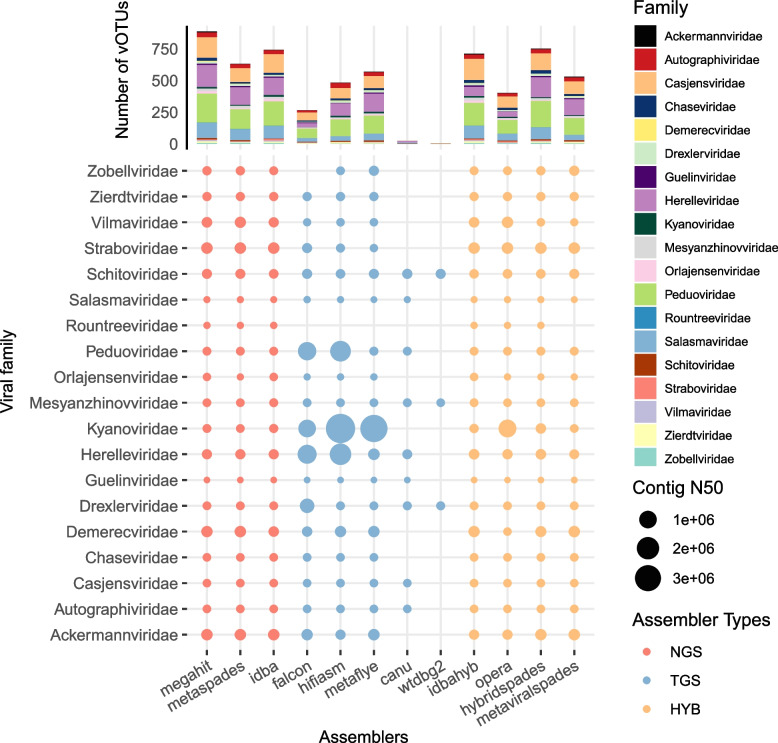
We evaluated the efficiency of NGS, TGS, and hybrid assemblers for viral genome discovery using 95 viral-like particle (VLP)-enriched fecal samples sequenced on both Illumina and PacBio platforms. MEGAHIT, metaFlye, and hybridSPAdes emerged as the optimal choices for NGS, TGS, and hybrid datasets, respectively. Notably, these assemblers recovered distinct viral genomes, demonstrating a remarkable degree of complementarity. By combining individual assembler results, we expanded the total number of nonredundant high-quality viral genomes by 4.83 ~ 21.7-fold compared to individual assemblers. Among them, viral genomes from NGS and TGS data have the least overlap, indicating the impact of data type on viral genome recovery. We also evaluated four binning methods, finding that CONCOCT incorporated more unrelated contigs into the same bins, while MetaBAT2, AVAMB, and vRhyme balanced inclusiveness and taxonomic consistency within bins.
Our findings highlight the challenges in metagenome-driven viral discovery, underscoring tool limitations. We advocate for combined use of multiple assemblers and sequencing technologies when feasible and highlight the urgent need for specialized tools tailored to gut virome assembly. This study contributes essential insights for advancing viral genome research in the context of gut metagenomics. Video Abstract.
宏基因组组装的病毒基因组极大地推动了人类肠道病毒组的发现和特征描述。然而,我们缺乏对组装工具在病毒基因组识别效率方面的比较评估,尤其是在下一代测序(NGS)和第三代测序(TGS)数据方面。
我们使用在Illumina和PacBio平台上测序的95个富含病毒样颗粒(VLP)的粪便样本,评估了NGS、TGS和混合组装器在病毒基因组发现方面的效率。MEGAHIT、metaFlye和hybridSPAdes分别成为NGS、TGS和混合数据集的最佳选择。值得注意的是,这些组装器恢复了不同的病毒基因组,显示出显著的互补程度。通过组合各个组装器的结果,与单个组装器相比,我们将非冗余高质量病毒基因组的总数扩大了4.83至21.7倍。其中,NGS和TGS数据中的病毒基因组重叠最少,表明数据类型对病毒基因组恢复的影响。我们还评估了四种分箱方法,发现CONCOCT将更多不相关的重叠群纳入同一箱中,而MetaBAT2、AVAMB和vRhyme在箱内平衡了包容性和分类一致性。
我们的研究结果突出了宏基因组驱动的病毒发现中的挑战,强调了工具的局限性。我们主张在可行时联合使用多种组装器和测序技术,并强调迫切需要针对肠道病毒组组装的专门工具。这项研究为在肠道宏基因组学背景下推进病毒基因组研究提供了重要见解。视频摘要。