Escorihuela-Sayalero Carlos, Sanuy Ares, Pardo Luis Carlos, Cazorla Claudio
Group of Characterization of Materials, Departament de Física, Universitat Politècnica de Catalunya, Campus Diagonal-Besòs, Av. Eduard Maristany 10-14, Barcelona 08019, Spain.
Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya, Campus Diagonal-Besòs, Av. Eduard Maristany 10-14, Barcelona 08019, Spain.
ACS Appl Mater Interfaces. 2025 Jan 8;17(1):1428-1440. doi: 10.1021/acsami.4c12762. Epub 2024 Dec 24.
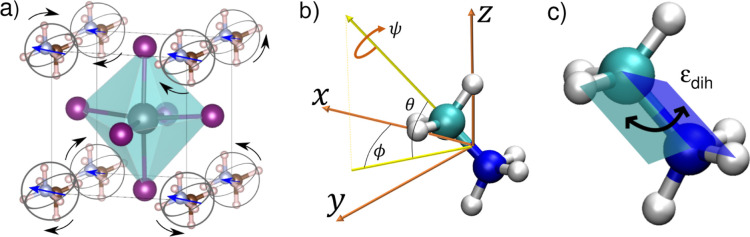
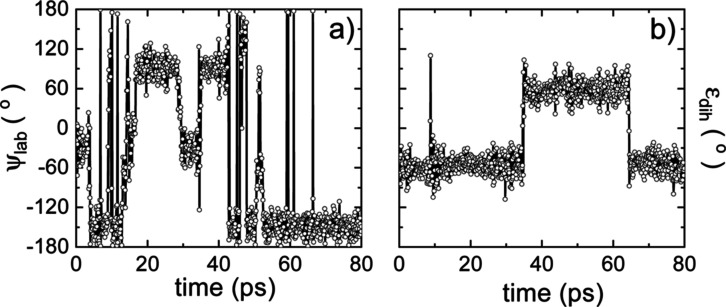
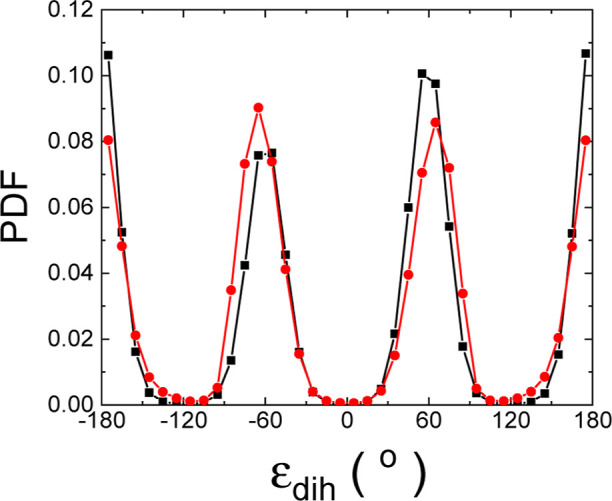
Hybrid organic-inorganic perovskites (HOIP) have emerged in recent years as highly promising semiconducting materials for a wide range of optoelectronic and energy applications. Nevertheless, the rotational dynamics of the organic components and many-molecule interdependencies, which may strongly impact the functional properties of HOIP, are not yet fully understood. In this study, we quantitatively analyze the orientational disorder and molecular correlations in archetypal perovskite CHNHPbI (MAPI) by performing comprehensive molecular dynamics simulations and entropy calculations. We found that, in addition to the usual vibrational and orientational contributions, rigid molecular rotations around the C-N axis and correlations between neighboring molecules noticeably contribute to the entropy increment associated with the temperature-induced order-disorder phase transition, Δ. Molecular conformational changes are equally infrequent in the low- ordered and high- disordered phases and have a null effect on Δ. Conversely, the couplings between the angular and vibrational degrees of freedom are substantially reinforced in the high- disordered phase and significantly counteract the phase-transition entropy increase resulting from other factors. Furthermore, the tendency for neighboring molecules to be orientationally ordered is markedly local, consequently inhibiting the formation of extensive polar nanodomains at both low and high temperatures. This theoretical investigation not only advances the fundamental knowledge of HOIP but also establishes physically insightful connections with contemporary technological applications like photovoltaics, solid-state cooling, and energy storage.
近年来,有机-无机杂化钙钛矿(HOIP)作为一类极具潜力的半导体材料,在广泛的光电子和能源应用领域崭露头角。然而,有机组分的旋转动力学以及多分子间的相互依赖性,可能会对HOIP的功能特性产生强烈影响,目前尚未得到充分理解。在本研究中,我们通过进行全面的分子动力学模拟和熵计算,定量分析了典型钙钛矿CHNHPbI(MAPI)中的取向无序和分子相关性。我们发现,除了通常的振动和取向贡献外,围绕C-N轴的刚性分子旋转以及相邻分子之间的相关性,对与温度诱导的有序-无序相变相关的熵增量Δ有显著贡献。分子构象变化在低序相和高无序相中同样不常见,并且对Δ没有影响。相反,在高无序相中,角自由度和振动自由度之间的耦合显著增强,并显著抵消了其他因素导致的相变熵增加。此外,相邻分子取向有序的趋势明显是局部的,因此在低温和高温下均抑制了广泛的极性纳米域的形成。这项理论研究不仅推进了对HOIP的基础知识的了解,还与光伏、固态冷却和能量存储等当代技术应用建立了具有物理洞察力的联系。