Xu Shuai, Jiang Jialiu, Chang Leilei, Zhang Biao, Zhu Xiaolei, Niu Fengnan
Department of Neurology, Nanjing Drum Tower Hospital, Affiliated Hospital of Medical School, Nanjing University, Zhongshan Road 321#, Nanjing, 210008, Jiangsu, China.
Department of Pathology, Nanjing Drum Tower Hospital, Affiliated Hospital of Medical School, Nanjing University, Zhongshan Road 321#, Nanjing, 210008, Jiangsu, China.
Orphanet J Rare Dis. 2024 Dec 24;19(1):487. doi: 10.1186/s13023-024-03511-4.
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a maternally inherited mitochondrial disorder that mostly affects the central nervous system and skeletal muscle. This study provides a comprehensive summary of the clinical symptoms, multisystemic pathogenesis, and genetic characteristics of MELAS syndrome. The aim was to improve comprehension of clinical practice and gain a deeper understanding of the latest pathophysiological theories.
The present investigation involved a cohort of patients diagnosed with MELAS at Nanjing Drum Tower Hospital between January 2014 and December 2022. Multisystem symptoms, magnetic resonance imaging/spectroscopy (MRI/MRS), muscle biopsy, and mitochondrial DNA (mtDNA) data were summarized and subsequently analysed.
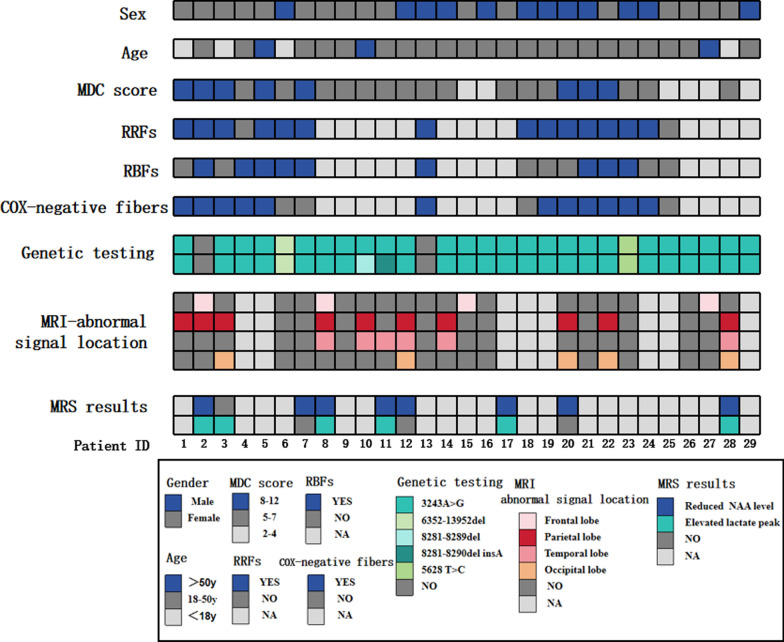
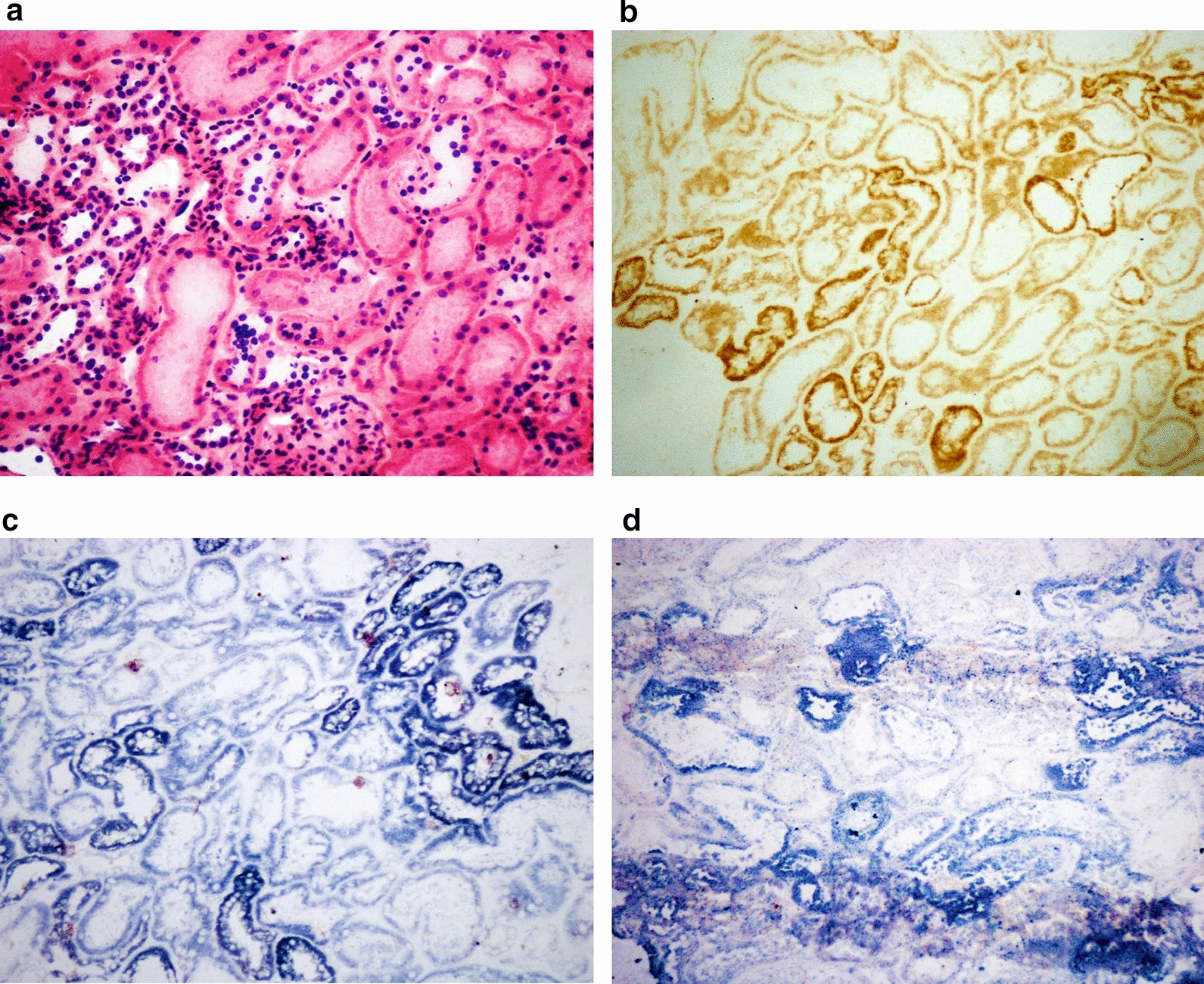
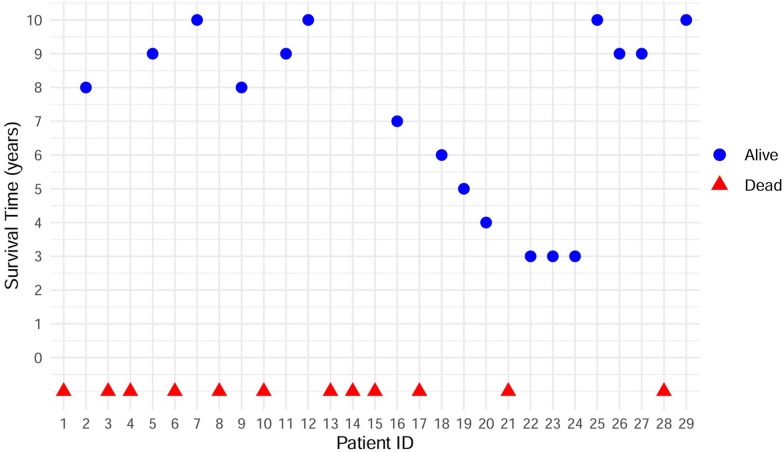
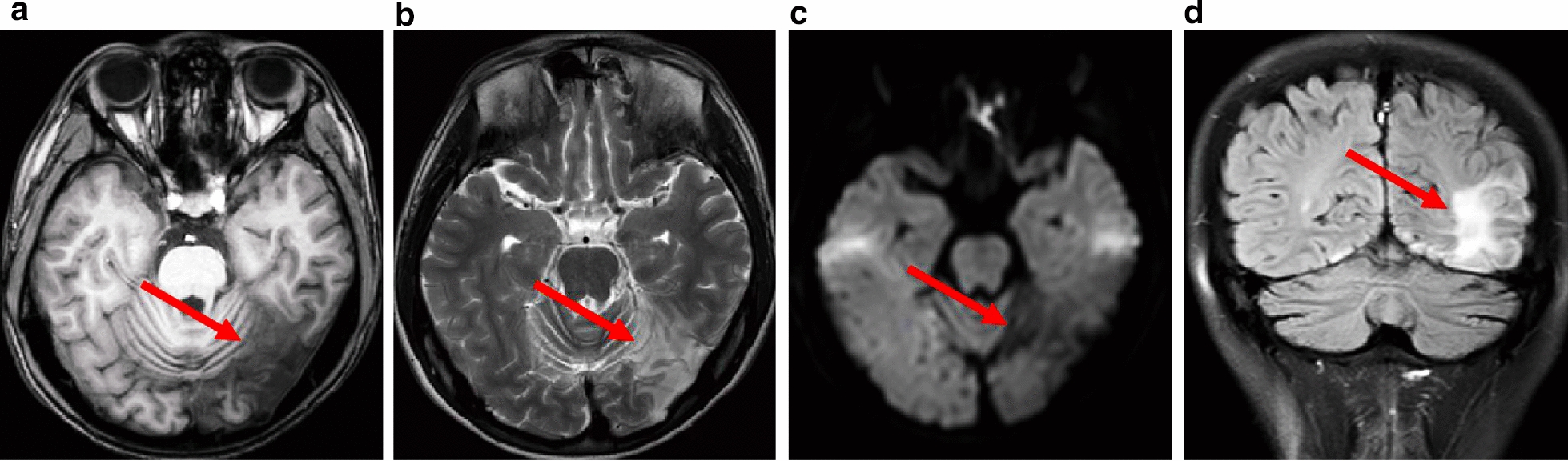
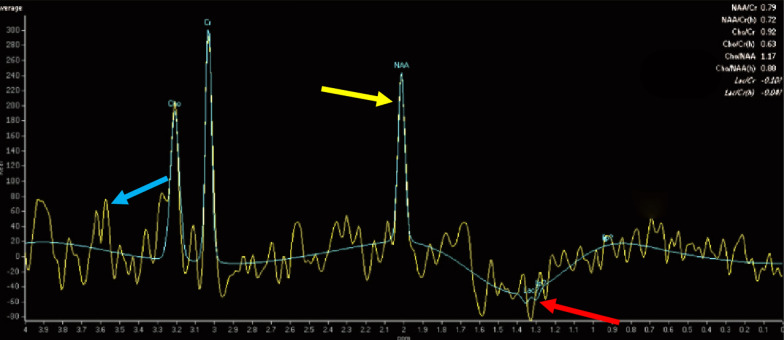
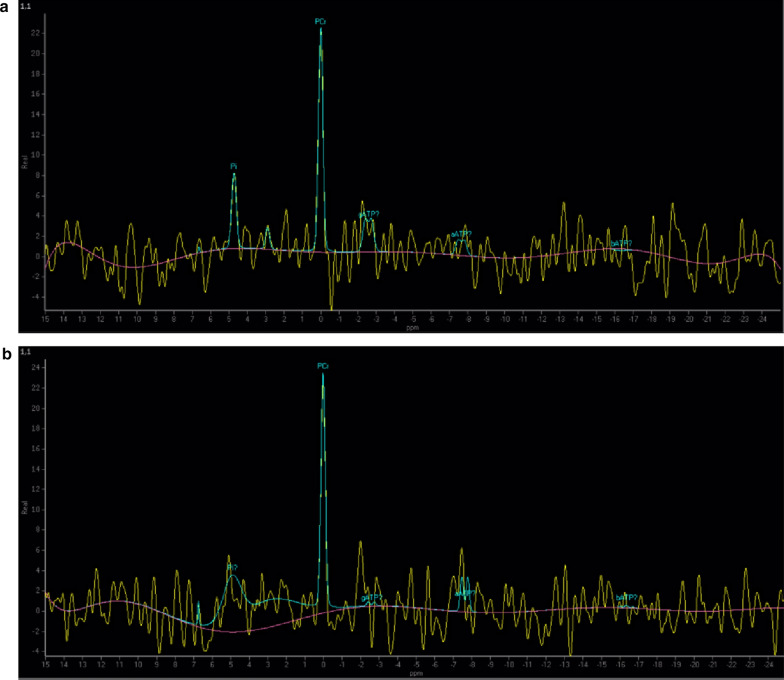
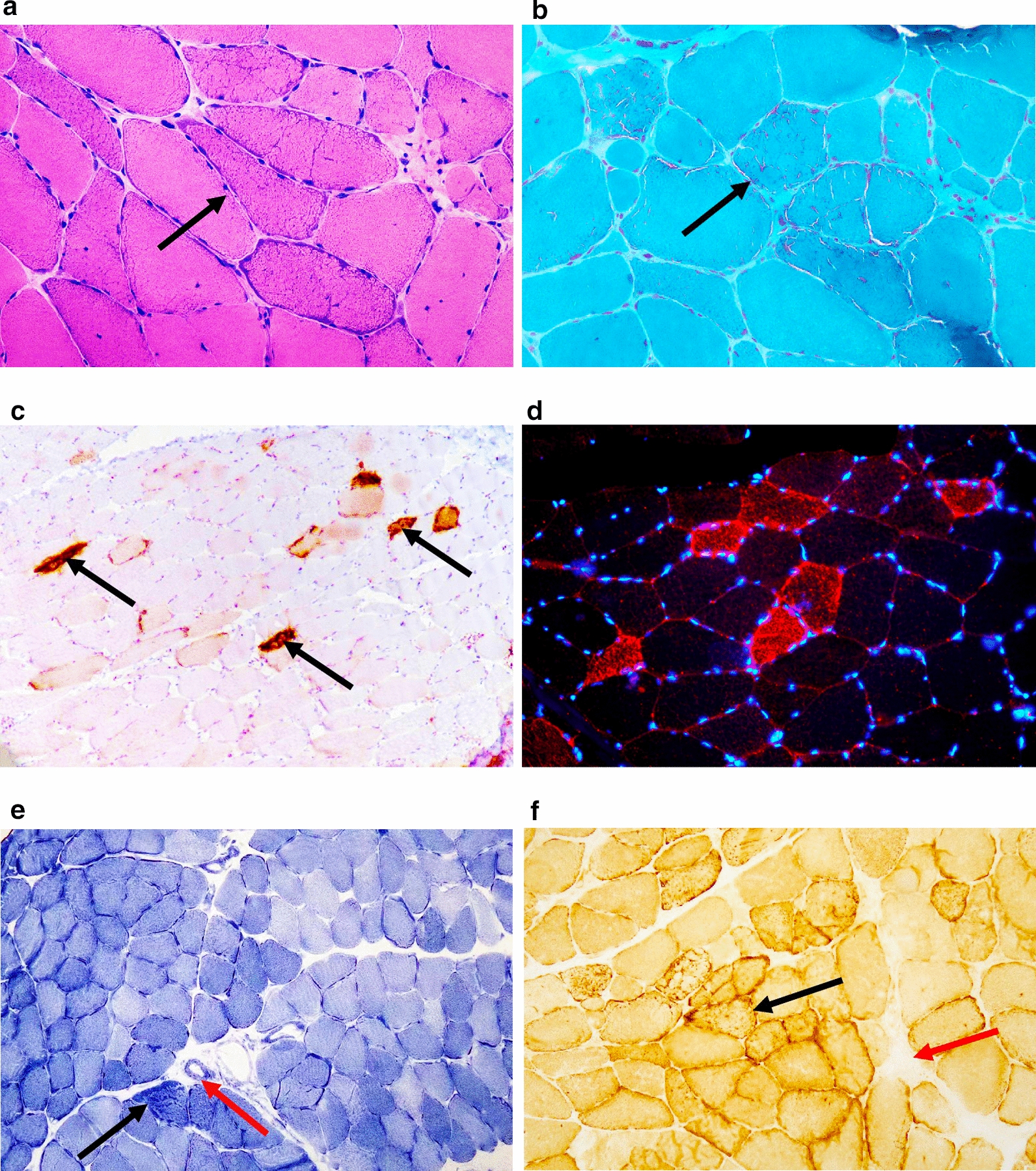
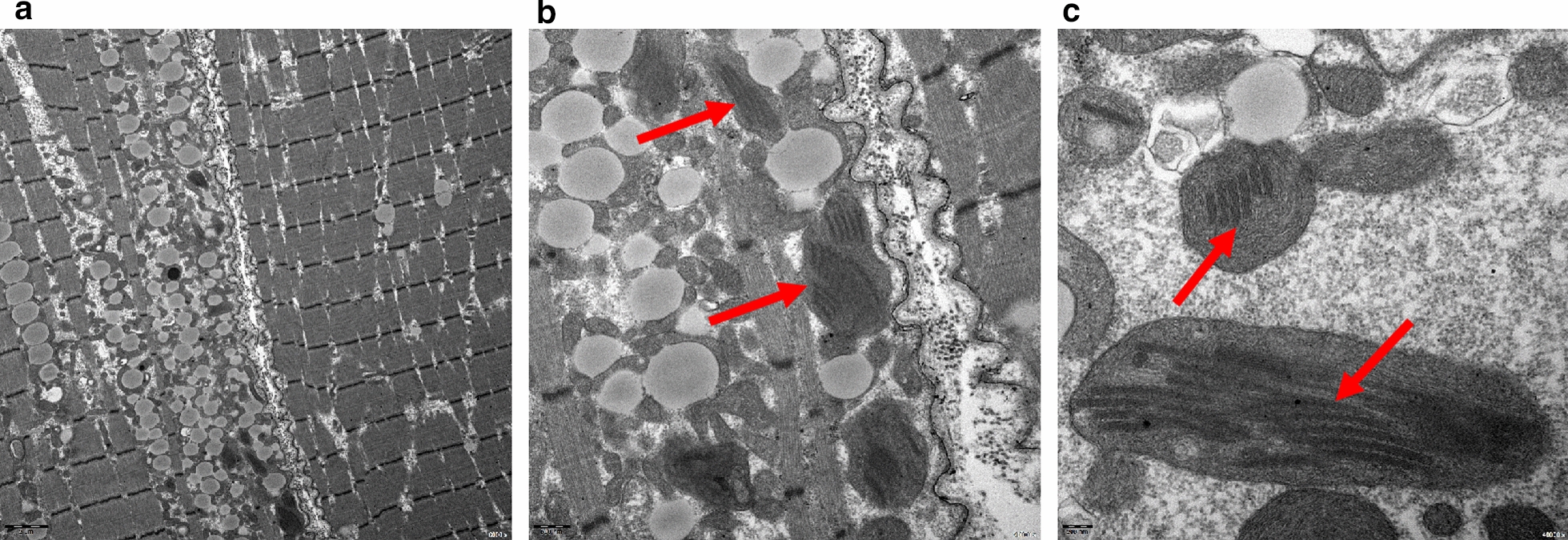
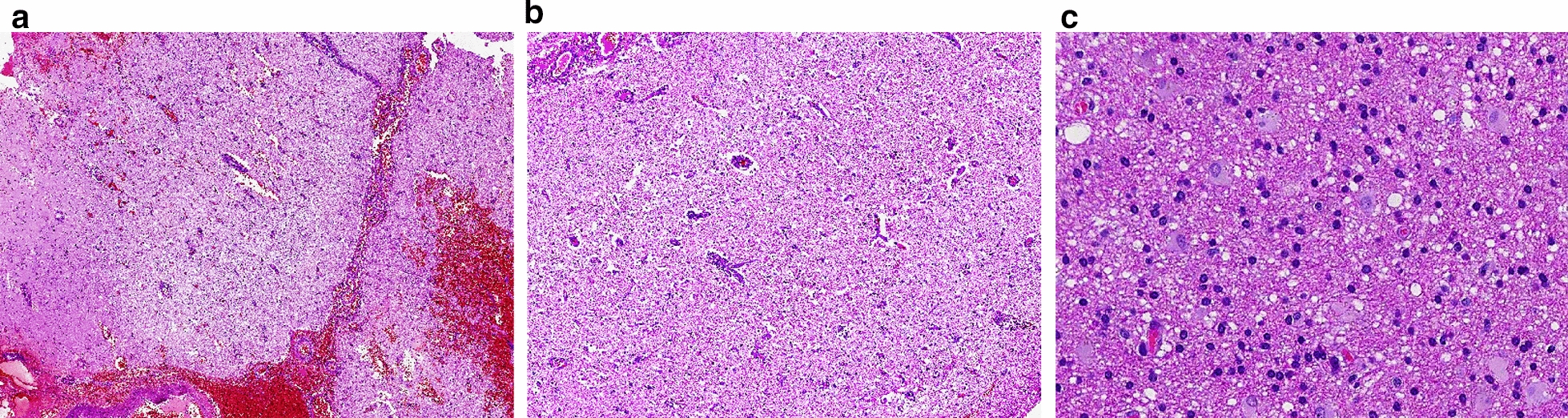
This retrospective study included a cohort of 29 MELAS patients who predominantly presented symptoms such as stroke-like episodes, proximal muscle weakness, and exercise intolerance. MRI scans revealed very small infarcts beneath the deep cortex during stroke-like episodes, indicating nonvascular brain damage. Pathology analyses of the brain also showed neuronal degeneration and glial cell proliferation in the cerebral parenchyma. Proton magnetic resonance spectroscopy (H-MRS) analysis revealed an increase in the lactate peak and a reduction in the N-acetylaspartate (NAA) level. Similarly, the phosphorus magnetic resonance spectroscopy (P-MRS) analysis revealed an abnormal ratio of inorganic phosphate (Pi) to phosphocreatine (PCr). Muscle biopsy revealed the presence of ragged red fibres (RRFs) and cytochrome c oxidase (COX) enzyme-defective cells. These abnormalities indicate structural abnormalities in the mitochondria and deficiencies in oxidative phosphorylation, respectively. In addition to the common m.3243A > G variant, other prevalent variants, including m.5628 T > C, m.6352-13952del, and a 9-bp small deletion combined with m.3243A > G, exist.
MELAS is a rare mitochondrial syndrome characterized by clinical heterogeneity and genetic heteroplasmy. Abnormalities in mitochondrial metabolic function and impairments in enzyme activity are the pathogenic processes underlying MELAS. Mitochondrial vasculopathy and mitochondrial neuropathy may provide a partial explanation for the unique aetiology of stroke-like episodes.
伴乳酸性酸中毒和卒中样发作的线粒体脑肌病(MELAS)综合征是一种母系遗传的线粒体疾病,主要影响中枢神经系统和骨骼肌。本研究全面总结了MELAS综合征的临床症状、多系统发病机制及遗传特征。目的是提高对临床实践的理解,并更深入地了解最新的病理生理理论。
本研究纳入了2014年1月至2022年12月期间在南京鼓楼医院确诊为MELAS的一组患者。总结并随后分析了多系统症状、磁共振成像/波谱分析(MRI/MRS)、肌肉活检及线粒体DNA(mtDNA)数据。
这项回顾性研究纳入了29例MELAS患者,主要表现为卒中样发作、近端肌无力和运动不耐受等症状。MRI扫描显示在卒中样发作期间,大脑深部皮质下有非常小的梗死灶,提示非血管性脑损伤。大脑病理分析还显示脑实质内神经元变性和胶质细胞增殖。质子磁共振波谱(H-MRS)分析显示乳酸峰升高,N-乙酰天门冬氨酸(NAA)水平降低。同样,磷磁共振波谱(P-MRS)分析显示无机磷酸盐(Pi)与磷酸肌酸(PCr)的比例异常。肌肉活检显示存在破碎红纤维(RRFs)和细胞色素c氧化酶(COX)酶缺陷细胞。这些异常分别表明线粒体存在结构异常和氧化磷酸化存在缺陷。除了常见的m.3243A>G变异外,还存在其他常见变异,包括m.5628T>C、m.6352-13952del,以及与m.3243A>G合并的9个碱基对的小缺失。
MELAS是一种罕见的线粒体综合征,具有临床异质性和遗传异质性。线粒体代谢功能异常和酶活性受损是MELAS的致病过程。线粒体血管病变和线粒体神经病变可能为卒中样发作的独特病因提供部分解释。