Wright Nicholas J, Matsuoka Yutaka, Park Hyeri, He Wei, Webster Caroline G, Furutani Kenta, Fedor Justin G, McGinnis Aidan, Zhao Yiquan, Chen Ouyang, Bang Sangsu, Fan Ping, Spasojevic Ivan, Hong Jiyong, Ji Ru-Rong, Lee Seok-Yong
Department of Biochemistry, Duke University School of Medicine, Durham, NC, 27710, USA.
Center for Translational Pain Medicine, Department of Anesthesiology, Duke University School of Medicine, Durham, NC, 27710, USA.
Nat Commun. 2024 Dec 30;15(1):10738. doi: 10.1038/s41467-024-54914-7.
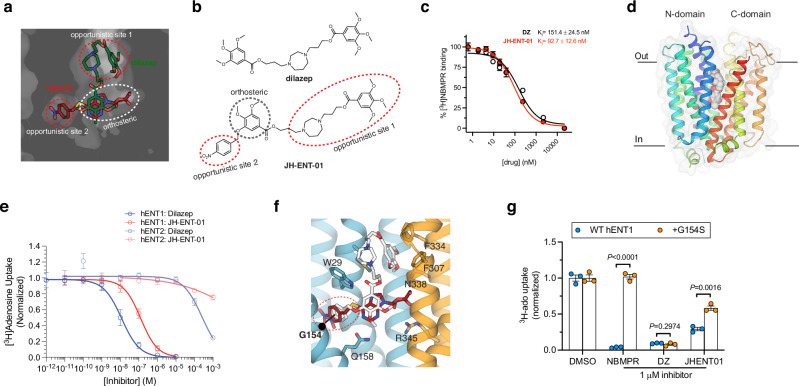
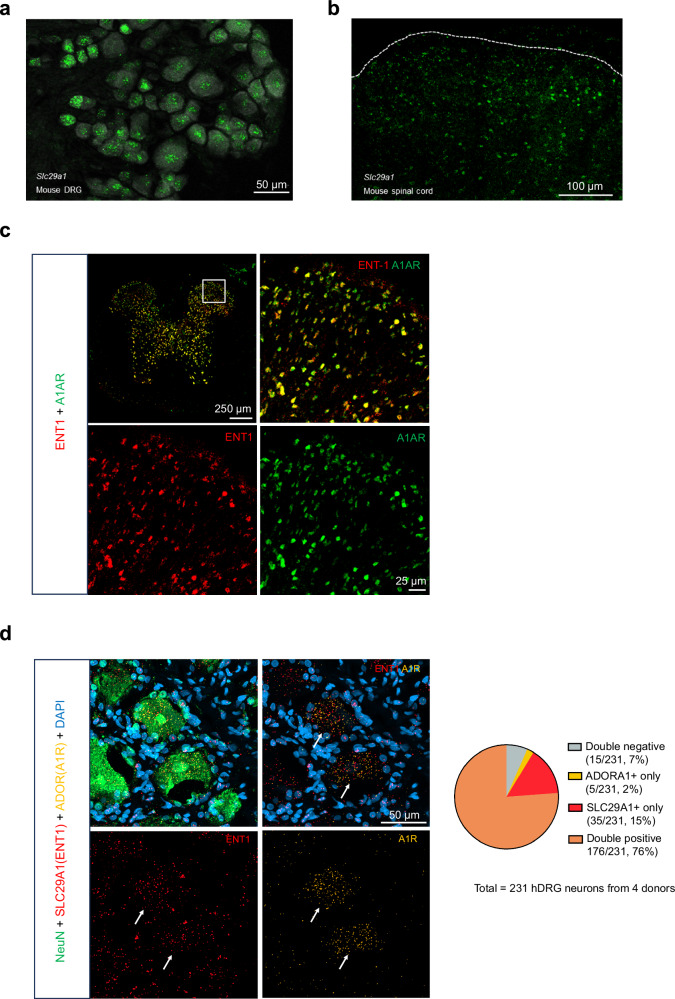
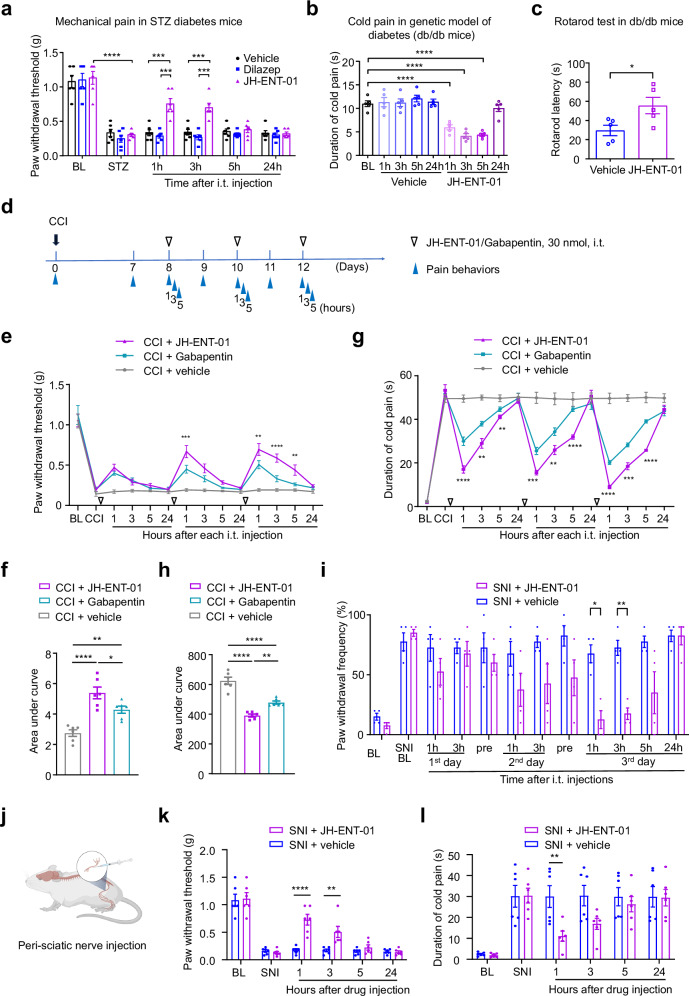
The current opioid crisis urgently calls for developing non-addictive pain medications. Progress has been slow, highlighting the need to uncover targets with unique mechanisms of action. Extracellular adenosine alleviates pain by activating the adenosine A1 receptor (A1R). However, efforts to develop A1R agonists have faced obstacles. The equilibrative nucleoside transporter subtype 1 (ENT1) plays a crucial role in regulating adenosine levels across cell membranes. We postulate that ENT1 inhibition may enhance extracellular adenosine levels, potentiating endogenous adenosine action at A1R and leading to analgesic effects. Here, we modify the ENT1 inhibitor dilazep based on its complex X-ray structure and show that this modified inhibitor reduces neuropathic and inflammatory pain in animal models while dilazep does not. Notably, our ENT1 inhibitor surpasses gabapentin in analgesic efficacy in a neuropathic pain model. Additionally, our inhibitor exhibits less cardiac side effect than dilazep via systemic administration and shows no side effects via local/intrathecal administration. ENT1 is colocalized with A1R in mouse and human dorsal root ganglia, and the analgesic effect of our inhibitor is linked to A1R. Our studies reveal ENT1 as a therapeutic target for analgesia, highlighting the promise of rationally designed ENT1 inhibitors for non-opioid pain medications.
当前的阿片类药物危机迫切需要研发非成瘾性止痛药物。进展一直缓慢,这凸显了发现具有独特作用机制靶点的必要性。细胞外腺苷通过激活腺苷A1受体(A1R)来减轻疼痛。然而,研发A1R激动剂的努力面临诸多障碍。平衡核苷转运体1型(ENT1)在调节细胞膜两侧的腺苷水平方面起着关键作用。我们推测,抑制ENT1可能会提高细胞外腺苷水平,增强内源性腺苷对A1R的作用并产生镇痛效果。在此,我们基于其复杂的X射线结构对ENT1抑制剂双嘧达莫进行修饰,结果表明这种修饰后的抑制剂能减轻动物模型中的神经性和炎性疼痛,而双嘧达莫则不能。值得注意的是,在神经性疼痛模型中,我们的ENT1抑制剂在镇痛效果上超过了加巴喷丁。此外,通过全身给药,我们的抑制剂比双嘧达莫表现出更少的心脏副作用,并且通过局部/鞘内给药未显示出副作用。在小鼠和人类背根神经节中,ENT1与A1R共定位,我们抑制剂的镇痛作用与A1R相关。我们的研究揭示ENT1是一个镇痛治疗靶点,突出了合理设计的ENT1抑制剂用于非阿片类止痛药物的前景。