Saelens Wouter, Pushkarev Olga, Deplancke Bart
Laboratory of Systems Biology and Genetics, Institute of Bio-engineering and Global Health Institute, School of Life Sciences, Swiss Federal Institute of Technology (EPFL), Lausanne, Switzerland.
Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Nat Commun. 2025 Jan 2;16(1):317. doi: 10.1038/s41467-024-55447-9.
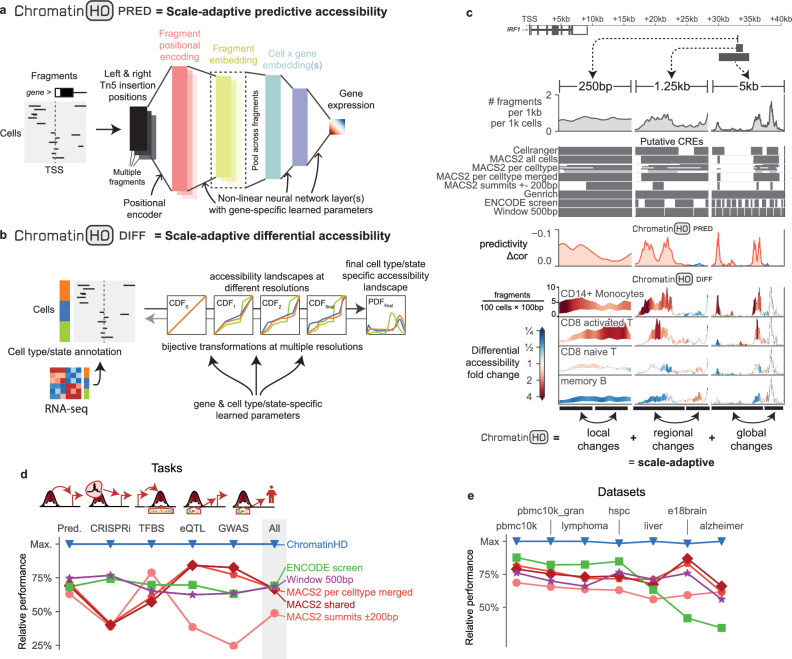
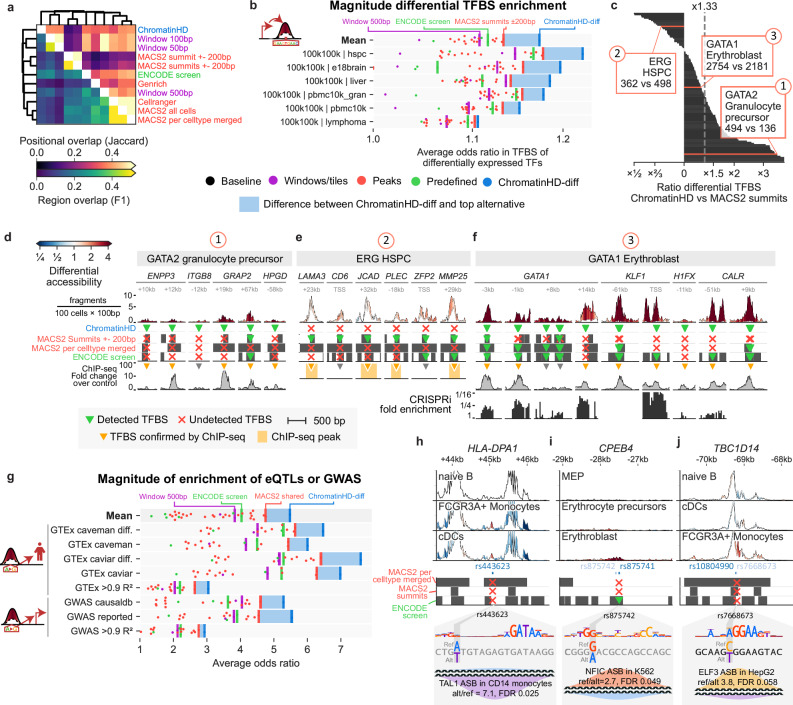
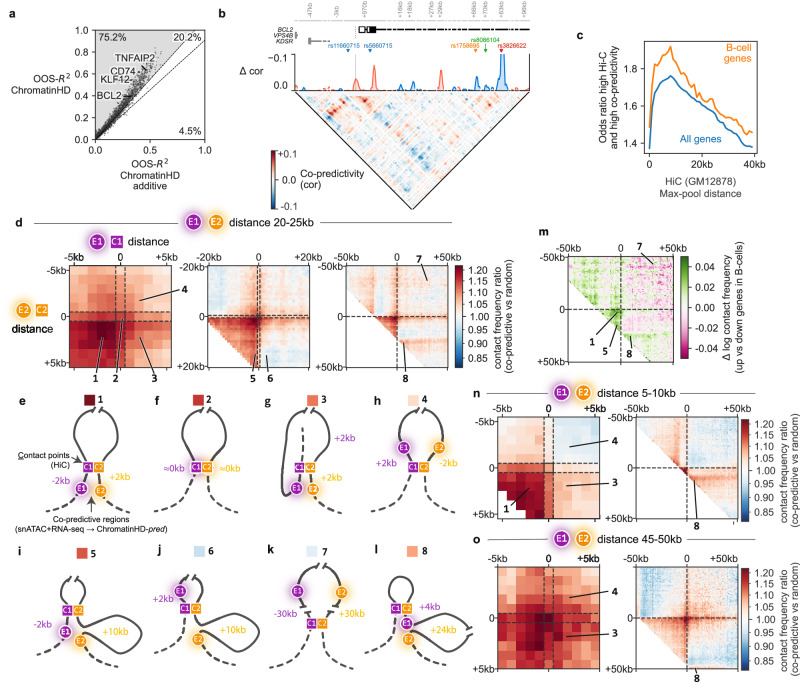
Gene regulation is inherently multiscale, but scale-adaptive machine learning methods that fully exploit this property in single-nucleus accessibility data are still lacking. Here, we develop ChromatinHD, a pair of scale-adaptive models that uses the raw accessibility data, without peak-calling or windows, to link regions to gene expression and determine differentially accessible chromatin. We show how ChromatinHD consistently outperforms existing peak and window-based approaches and find that this is due to a large number of uniquely captured, functional accessibility changes within and outside of putative cis-regulatory regions. Furthermore, ChromatinHD can delineate collaborating regulatory regions, including their preferential genomic conformations, that drive gene expression. Finally, our models also use changes in ATAC-seq fragment lengths to identify dense binding of transcription factors, a feature not captured by footprinting methods. Altogether, ChromatinHD, available at https://chromatinhd.org , is a suite of computational tools that enables a data-driven understanding of chromatin accessibility at various scales and how it relates to gene expression.
基因调控本质上是多尺度的,但在单核可及性数据中充分利用这一特性的尺度自适应机器学习方法仍然缺乏。在这里,我们开发了ChromatinHD,这是一对尺度自适应模型,它使用原始可及性数据,无需进行峰调用或窗口划分,即可将区域与基因表达联系起来,并确定差异可及的染色质。我们展示了ChromatinHD如何始终优于现有的基于峰和窗口的方法,并发现这是由于在假定的顺式调控区域内外有大量独特捕获的功能性可及性变化。此外,ChromatinHD可以描绘出协同调控区域,包括它们优先的基因组构象,这些区域驱动基因表达。最后,我们的模型还利用ATAC-seq片段长度的变化来识别转录因子的密集结合,这是足迹法无法捕获的一个特征。总之,可在https://chromatinhd.org上获取的ChromatinHD是一套计算工具,能够实现对不同尺度下染色质可及性及其与基因表达关系的数据驱动理解。