Isei Michael O, Crockett Meredith, Chen Emily, Rodwell-Bullock Joel, Carroll Trae, Girardi Peter A, Nehrke Keith, Johnson Gail V W
Department of Anesthesiology & Perioperative Medicine, University of Rochester, Rochester, New York, United States of America.
Nephrology Division, Department of Medicine, University of Rochester, Rochester, New York, United States of America.
PLoS One. 2025 Jan 3;20(1):e0307358. doi: 10.1371/journal.pone.0307358. eCollection 2025.
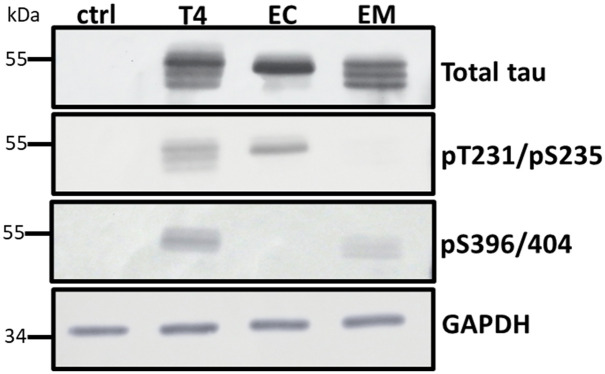
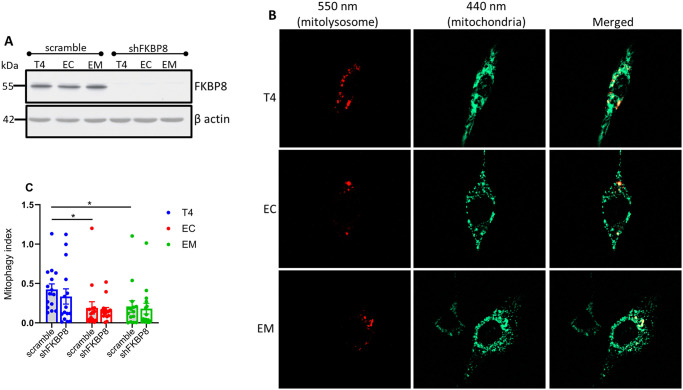
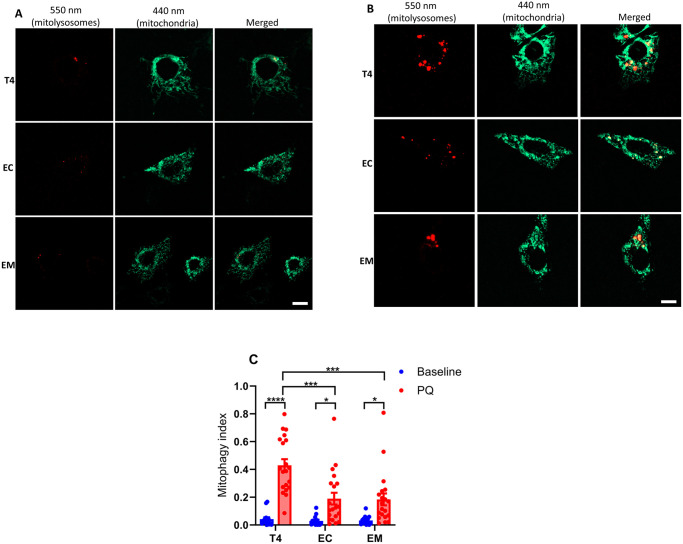
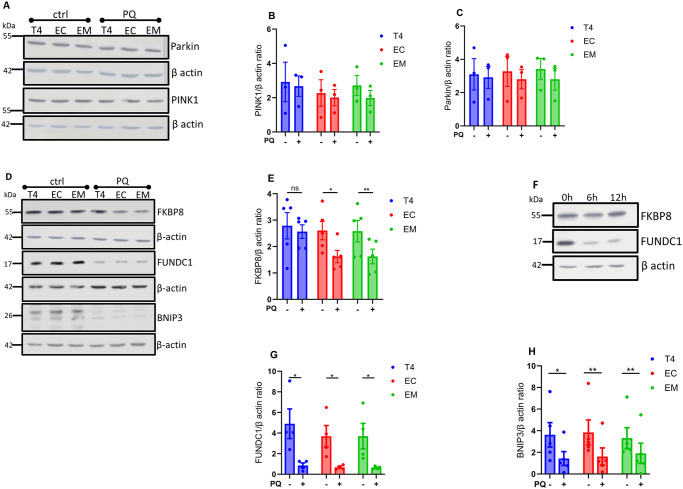
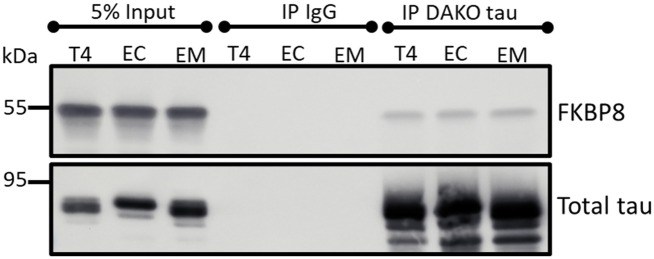
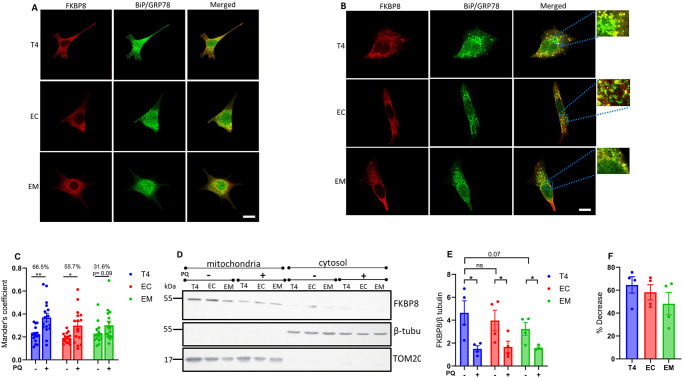
Neurodegenerative diseases are often characterized by mitochondrial dysfunction. In Alzheimer's disease, abnormal tau phosphorylation disrupts mitophagy, a quality control process through which damaged organelles are selectively removed from the mitochondrial network. The precise mechanism through which this occurs remains unclear. Previously, we showed that tau which has been mutated at Thr-231 to glutamic acid to mimic an Alzheimer's-relevant phospho-epitope expressed early in disease selectively inhibits oxidative stress-induced mitophagy in Caenorhabditis elegans. Here, we use immortalized mouse hippocampal neuronal cell lines to extend that result into mammalian cells. Specifically, we show that phosphomimetic tau at Ser-396/404 (EC) or Thr-231/Ser-235 (EM) partly inhibits mitophagy induction by paraquat, a potent inducer of mitochondrial oxidative stress. Moreover, a combination of immunologic and biochemical approaches demonstrates that the levels of the mitophagy receptor FKBP8, significantly decrease in response to paraquat in cells expressing EC or EM tau mutants, but not in cells expressing wildtype tau. In contrast, paraquat treatment results in a decrease in the levels of the mitophagy receptors FUNDC1 and BNIP3 in the presence of both wildtype tau and the tau mutants. Interestingly, FKBP8 is normally trafficked to the endoplasmic reticulum during oxidative stress induced mitophagy, and our results support a model where this trafficking is impacted by disease-relevant tau, perhaps through a direct interaction. We provide new insights into the molecular mechanisms underlying tau pathology in Alzheimer's disease and highlight FKBP8 receptor as a potential target for mitigating mitochondrial dysfunction in neurodegenerative diseases.
神经退行性疾病通常以线粒体功能障碍为特征。在阿尔茨海默病中,异常的tau蛋白磷酸化会破坏线粒体自噬,这是一种质量控制过程,受损细胞器会通过该过程从线粒体网络中被选择性清除。其发生的确切机制尚不清楚。此前,我们发现将苏氨酸-231突变为谷氨酸以模拟疾病早期出现的与阿尔茨海默病相关的磷酸化表位的tau蛋白,会选择性抑制秀丽隐杆线虫中氧化应激诱导的线粒体自噬。在此,我们使用永生化小鼠海马神经元细胞系将该结果扩展到哺乳动物细胞。具体而言,我们发现丝氨酸-396/404(EC)或苏氨酸-231/丝氨酸-235(EM)处的磷酸化模拟tau蛋白部分抑制百草枯诱导的线粒体自噬,百草枯是线粒体氧化应激的强效诱导剂。此外,免疫和生化方法相结合表明,在表达EC或EM tau突变体的细胞中,线粒体自噬受体FKBP8的水平会因百草枯而显著降低,但在表达野生型tau的细胞中则不会。相比之下,在存在野生型tau和tau突变体的情况下,百草枯处理会导致线粒体自噬受体FUNDC1和BNIP3的水平降低。有趣的是,在氧化应激诱导的线粒体自噬过程中,FKBP8通常会被转运到内质网,我们的结果支持这样一种模型,即这种转运受到与疾病相关的tau蛋白的影响,可能是通过直接相互作用。我们为阿尔茨海默病中tau蛋白病理的分子机制提供了新的见解,并强调FKBP8受体是减轻神经退行性疾病中线粒体功能障碍的潜在靶点。