Majidian Sina, Nevers Yannis, Yazdizadeh Kharrazi Ali, Warwick Vesztrocy Alex, Pascarelli Stefano, Moi David, Glover Natasha, Altenhoff Adrian M, Dessimoz Christophe
Department of Computational Biology, University of Lausanne, Lausanne, Switzerland.
Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Nat Methods. 2025 Feb;22(2):269-272. doi: 10.1038/s41592-024-02552-8. Epub 2025 Jan 3.
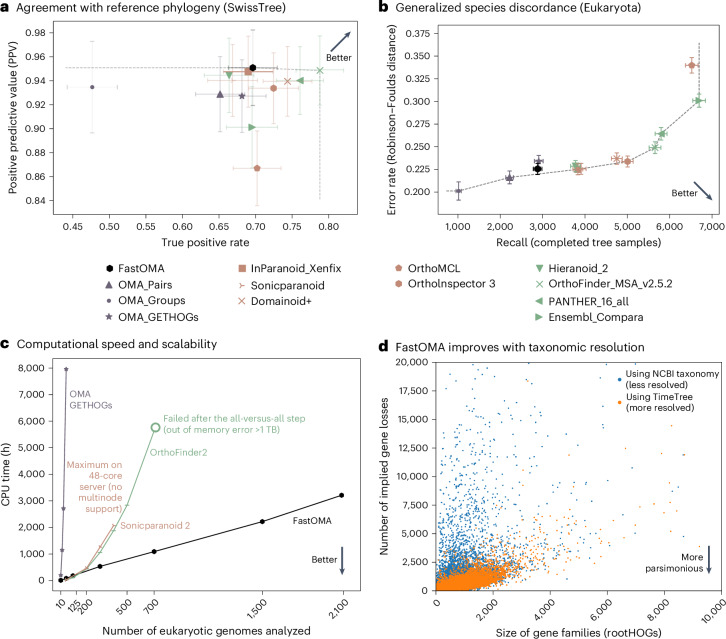
The surge in genome data, with ongoing efforts aiming to sequence 1.5 M eukaryotes in a decade, could revolutionize genomics, revealing the origins, evolution and genetic innovations of biological processes. Yet, traditional genomics methods scale poorly with such large datasets. Here, addressing this, 'FastOMA' provides linear scalability for orthology inference, enabling the processing of thousands of eukaryotic genomes within a day. FastOMA maintains the high accuracy and resolution of the well-established Orthologous Matrix (OMA) approach in benchmarks. FastOMA is available via GitHub at https://github.com/DessimozLab/FastOMA/ .
基因组数据激增,目前正努力在十年内对150万个真核生物进行测序,这可能会彻底改变基因组学,揭示生物过程的起源、进化和遗传创新。然而,传统的基因组学方法在处理如此庞大的数据集时效果不佳。在此,为解决这一问题,“FastOMA”为直系同源推断提供了线性可扩展性,能够在一天内处理数千个真核生物基因组。在基准测试中,FastOMA保持了成熟的直系同源矩阵(OMA)方法的高精度和分辨率。可通过GitHub上的https://github.com/DessimozLab/FastOMA/获取FastOMA。