Chu Simon N, Soupene Eric, Sharma Devesh, Sinha Roshani, McCreary Travis, Hernandez Britney, Shen Huifeng, Wienert Beeke, Bowman Chance, Yin Han, Lesch Benjamin J, Jia Kun, Romero Kathleen A, Kostamo Zachary, Zhang Yankai, Tran Tammy, Cordero Marco, Homma Shota, Hampton Jessica P, Gardner James M, Conklin Bruce R, MacKenzie Tippi C, Sheehan Vivien A, Porteus Matthew H, Cromer M Kyle
Department of Surgery, University of California, San Francisco, San Francisco, CA 94143, USA; Eli & Edythe Broad Center for Regeneration Medicine, University of California, San Francisco, San Francisco, CA 94143, USA; Diabetes Center, University of California, San Francisco, San Francisco, CA 94143, USA.
Department of Pediatrics, University of California, San Francisco, Oakland, CA 94609, USA.
Cell Rep. 2025 Jan 28;44(1):115141. doi: 10.1016/j.celrep.2024.115141. Epub 2025 Jan 3.
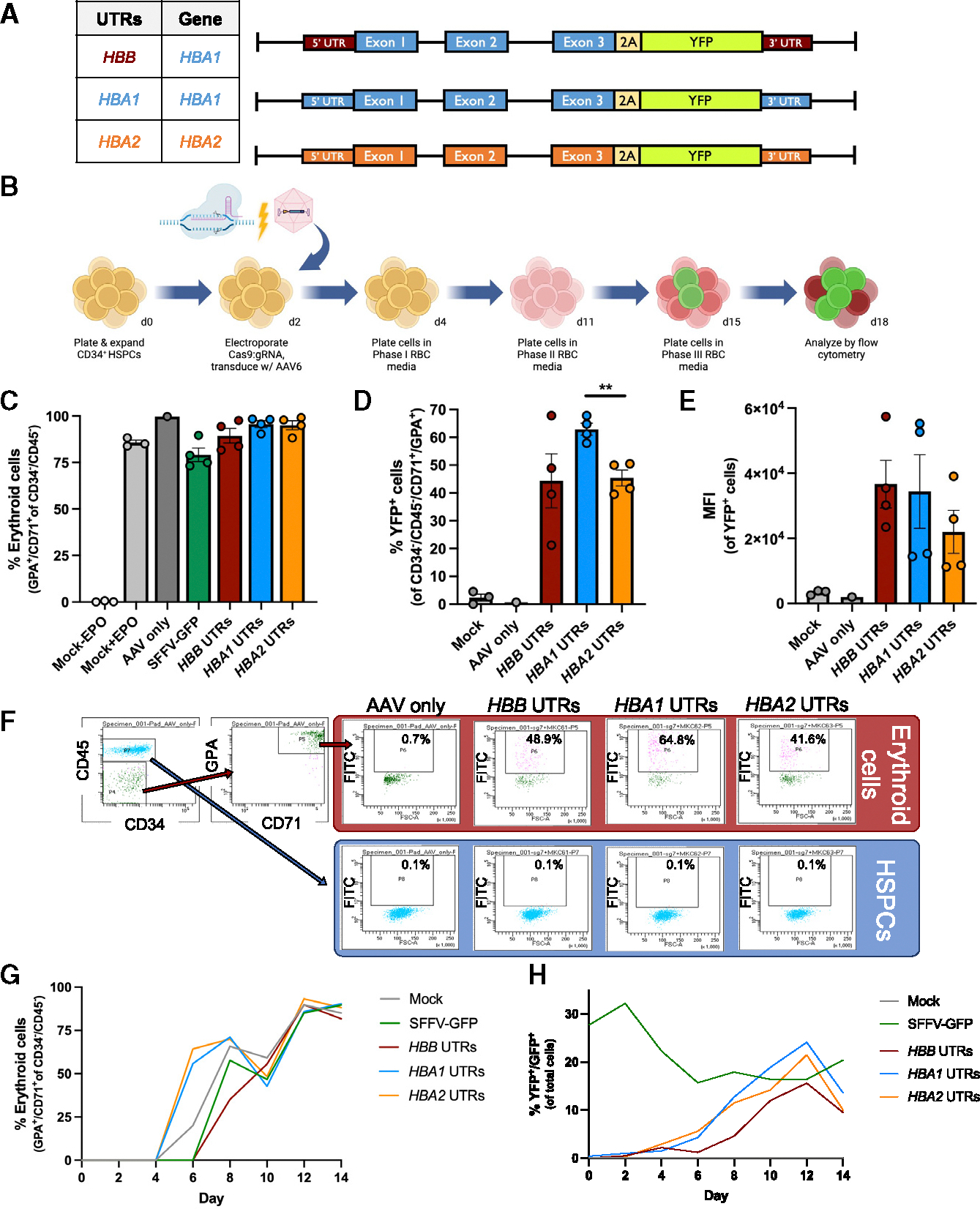
The most severe form of α-thalassemia results from loss of all four copies of α-globin. Postnatally, patients face challenges similar to β-thalassemia, including severe anemia and erythrotoxicity due to the imbalance of β-globin and α-globin chains. Despite progress in genome editing treatments for β-thalassemia, there is no analogous curative option for α-thalassemia. To address this, we designed a Cas9/AAV6-mediated genome editing strategy that integrates a functional α-globin gene into the β-globin locus in α-thalassemia patient-derived hematopoietic stem and progenitor cells (HSPCs). Incorporation of a truncated erythropoietin receptor transgene into the α-globin integration cassette significantly increased erythropoietic output from edited HSPCs and led to the most robust production of α-globin, and consequently hemoglobin tetramers. By directing edited HSPCs toward increased production of clinically relevant erythroid cells, this approach has the potential to mitigate the limitations of current treatments for the hemoglobinopathies, including low genome editing and low engraftment rates.
α地中海贫血最严重的形式是由于α珠蛋白的所有四个拷贝缺失所致。出生后,患者面临与β地中海贫血类似的挑战,包括由于β珠蛋白和α珠蛋白链失衡导致的严重贫血和红细胞毒性。尽管β地中海贫血的基因组编辑治疗取得了进展,但对于α地中海贫血尚无类似的治愈方案。为了解决这一问题,我们设计了一种Cas9/AAV6介导的基因组编辑策略,将一个功能性α珠蛋白基因整合到α地中海贫血患者来源的造血干细胞和祖细胞(HSPCs)的β珠蛋白基因座中。将截短的促红细胞生成素受体转基因整合到α珠蛋白整合盒中,显著提高了编辑后的HSPCs的红细胞生成产量,并导致α珠蛋白以及血红蛋白四聚体的最强有力生成。通过引导编辑后的HSPCs增加临床相关红系细胞的生成,这种方法有可能减轻目前血红蛋白病治疗的局限性,包括低基因组编辑率和低植入率。