Department of Surgery, University of California, San Francisco, San Francisco, CA 94143, USA; Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, CA 94143, USA; Eli and Edythe Broad Center for Regeneration Medicine, University of California, San Francisco, San Francisco, CA 94143, USA.
Department of Pediatrics, Stanford University, Stanford, CA 94305, USA.
Mol Ther. 2023 Apr 5;31(4):1074-1087. doi: 10.1016/j.ymthe.2023.02.011. Epub 2023 Feb 15.
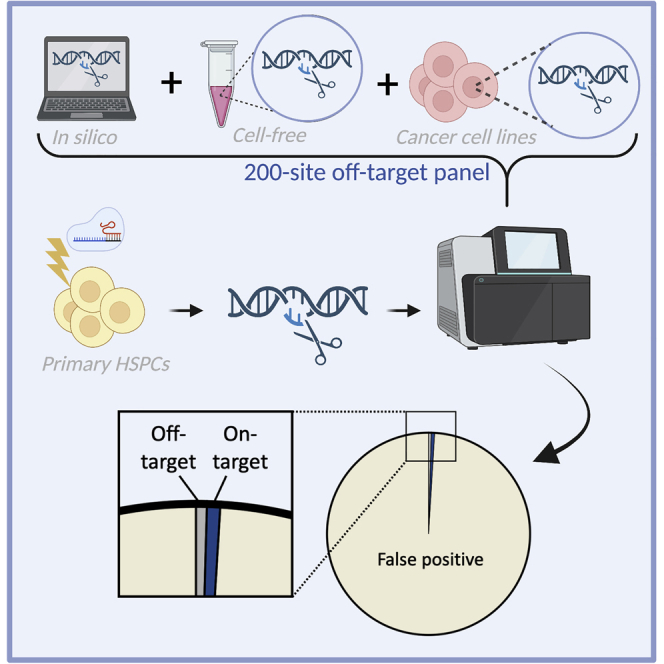
While a number of methods exist to investigate CRISPR off-target (OT) editing, few have been compared head-to-head in primary cells after clinically relevant editing processes. Therefore, we compared in silico tools (COSMID, CCTop, and Cas-OFFinder) and empirical methods (CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, and SITE-Seq) after ex vivo hematopoietic stem and progenitor cell (HSPC) editing. We performed editing using 11 different gRNAs complexed with Cas9 protein (high-fidelity [HiFi] or wild-type versions), then performed targeted next-generation sequencing of nominated OT sites identified by in silico and empirical methods. We identified an average of less than one OT site per guide RNA (gRNA) and all OT sites generated using HiFi Cas9 and a 20-nt gRNA were identified by all OT detection methods with the exception of SITE-seq. This resulted in high sensitivity for the majority of OT nomination tools and COSMID, DISCOVER-Seq, and GUIDE-Seq attained the highest positive predictive value (PPV). We found that empirical methods did not identify OT sites that were not also identified by bioinformatic methods. This study supports that refined bioinformatic algorithms could be developed that maintain both high sensitivity and PPV, thereby enabling more efficient identification of potential OT sites without compromising a thorough examination for any given gRNA.
虽然有许多方法可用于研究 CRISPR 脱靶(OT)编辑,但在临床相关编辑过程后,很少有方法在原代细胞中进行头对头比较。因此,我们在体外造血干细胞和祖细胞(HSPC)编辑后比较了计算工具(COSMID、CCTop 和 Cas-OFFinder)和经验方法(CHANGE-Seq、CIRCLE-Seq、DISCOVER-Seq、GUIDE-Seq 和 SITE-Seq)。我们使用 11 种不同的与 Cas9 蛋白复合的 gRNA(高保真 [HiFi] 或野生型)进行编辑,然后对通过计算和经验方法确定的提名 OT 位点进行靶向下一代测序。我们发现每个 gRNA 的平均 OT 位点少于一个,并且所有使用 HiFi Cas9 和 20nt gRNA 生成的 OT 位点都可以通过所有 OT 检测方法(除了 SITE-seq)识别。这导致大多数 OT 提名工具具有高灵敏度,并且 COSMID、DISCOVER-Seq 和 GUIDE-Seq 获得了最高的阳性预测值(PPV)。我们发现经验方法并未识别出生物信息学方法未识别的 OT 位点。这项研究支持可以开发出更精细的生物信息学算法,这些算法既能保持高灵敏度又能保持高阳性预测值,从而能够更有效地识别潜在的 OT 位点,而不会影响对任何特定 gRNA 的全面检查。