Integrated Genetic Approaches in Therapeutic Discovery for Rare Diseases (INTEGRARE), Genethon, Unité Mixte de Recherche (UMR) S951 INSERM, University Evry, University Paris-Saclay, Evry, France.
Imagine Institute, UMR 163 INSERM, Paris, France.
Blood Adv. 2021 Mar 9;5(5):1137-1153. doi: 10.1182/bloodadvances.2020001996.
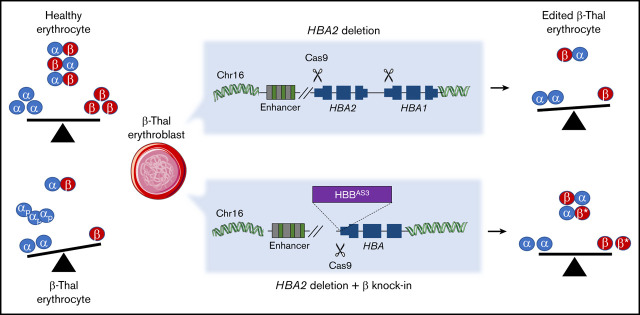
β-thalassemias (β-thal) are a group of blood disorders caused by mutations in the β-globin gene (HBB) cluster. β-globin associates with α-globin to form adult hemoglobin (HbA, α2β2), the main oxygen-carrier in erythrocytes. When β-globin chains are absent or limiting, free α-globins precipitate and damage cell membranes, causing hemolysis and ineffective erythropoiesis. Clinical data show that severity of β-thal correlates with the number of inherited α-globin genes (HBA1 and HBA2), with α-globin gene deletions having a beneficial effect for patients. Here, we describe a novel strategy to treat β-thal based on genome editing of the α-globin locus in human hematopoietic stem/progenitor cells (HSPCs). Using CRISPR/Cas9, we combined 2 therapeutic approaches: (1) α-globin downregulation, by deleting the HBA2 gene to recreate an α-thalassemia trait, and (2) β-globin expression, by targeted integration of a β-globin transgene downstream the HBA2 promoter. First, we optimized the CRISPR/Cas9 strategy and corrected the pathological phenotype in a cellular model of β-thalassemia (human erythroid progenitor cell [HUDEP-2] β0). Then, we edited healthy donor HSPCs and demonstrated that they maintained long-term repopulation capacity and multipotency in xenotransplanted mice. To assess the clinical potential of this approach, we next edited β-thal HSPCs and achieved correction of α/β globin imbalance in HSPC-derived erythroblasts. As a safer option for clinical translation, we performed editing in HSPCs using Cas9 nickase showing precise editing with no InDels. Overall, we described an innovative CRISPR/Cas9 approach to improve α/β globin imbalance in thalassemic HSPCs, paving the way for novel therapeutic strategies for β-thal.
β-地中海贫血症(β-thal)是一组由β-球蛋白基因(HBB)簇突变引起的血液疾病。β-球蛋白与α-球蛋白结合形成成人血红蛋白(HbA,α2β2),是红细胞中主要的氧载体。当β-球蛋白链缺失或不足时,游离的α-球蛋白沉淀并破坏细胞膜,导致溶血和无效的红细胞生成。临床数据表明,β-地中海贫血症的严重程度与遗传的α-球蛋白基因(HBA1 和 HBA2)数量相关,α-球蛋白基因缺失对患者有有益的影响。在这里,我们描述了一种基于人类造血干/祖细胞(HSPCs)α-球蛋白基因座基因组编辑治疗β-地中海贫血症的新策略。我们使用 CRISPR/Cas9 组合了 2 种治疗方法:(1)α-球蛋白下调,通过删除 HBA2 基因来重新创建α-地中海贫血特征,以及(2)β-球蛋白表达,通过靶向整合 HBA2 启动子下游的β-球蛋白转基因。首先,我们优化了 CRISPR/Cas9 策略,并在β-地中海贫血症的细胞模型(人类红系祖细胞[HUDEP-2]β0)中纠正了病理表型。然后,我们编辑了健康供体 HSPCs,并证明它们在异种移植小鼠中保持了长期的重建能力和多能性。为了评估这种方法的临床潜力,我们接下来编辑了β-地中海贫血症的 HSPCs,并在 HSPC 衍生的红细胞中实现了α/β 球蛋白失衡的纠正。作为临床转化的更安全选择,我们使用 Cas9 切口酶在 HSPCs 中进行编辑,显示出精确的编辑而没有插入缺失。总体而言,我们描述了一种创新性的 CRISPR/Cas9 方法,可改善地中海贫血症 HSPCs 中的α/β 球蛋白失衡,为β-地中海贫血症的新治疗策略铺平了道路。