Wang Xiao, Zhang Yuanyuan, Ray Suhita, Jha Anupama, Fang Tangqi, Hang Shengqi, Doulatov Sergei, Noble William Stafford, Wang Sheng
Department of Genome Sciences, University of Washington, Seattle, WA, USA.
Paul G. Allen School of Computer Science and Engineering, University of Washington, Seattle, WA, 98105, USA.
bioRxiv. 2024 Dec 20:2024.12.16.628821. doi: 10.1101/2024.12.16.628821.
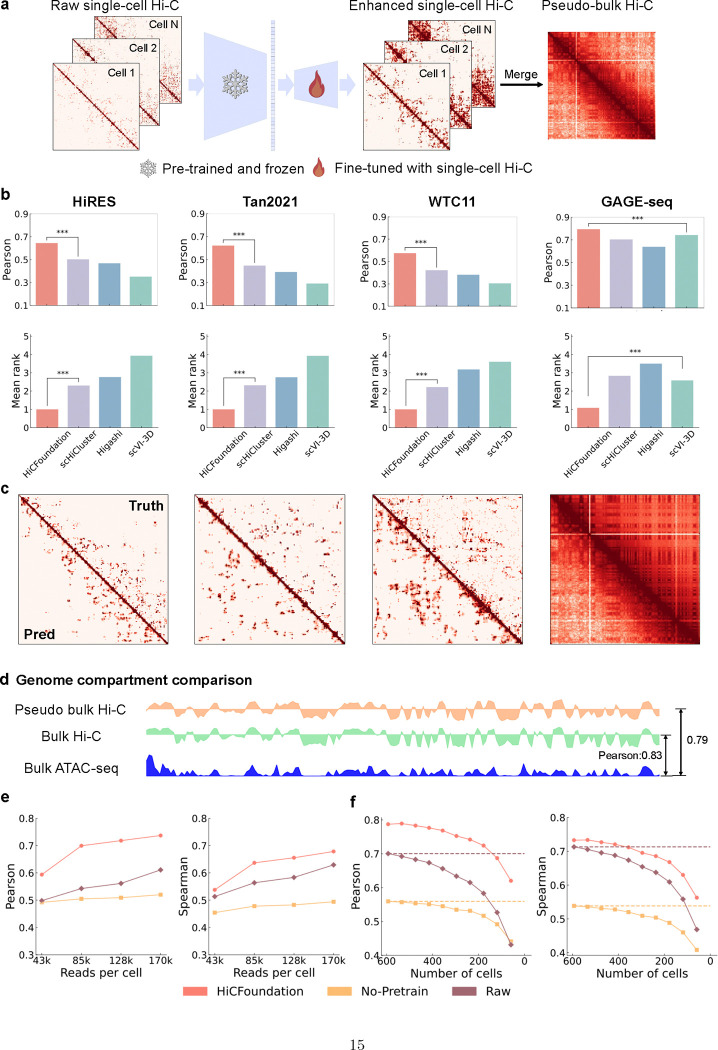
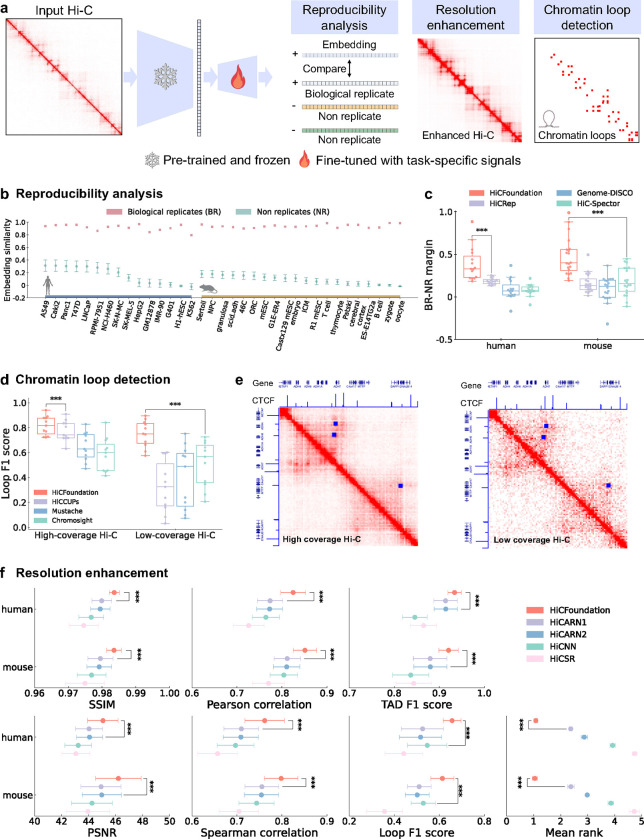
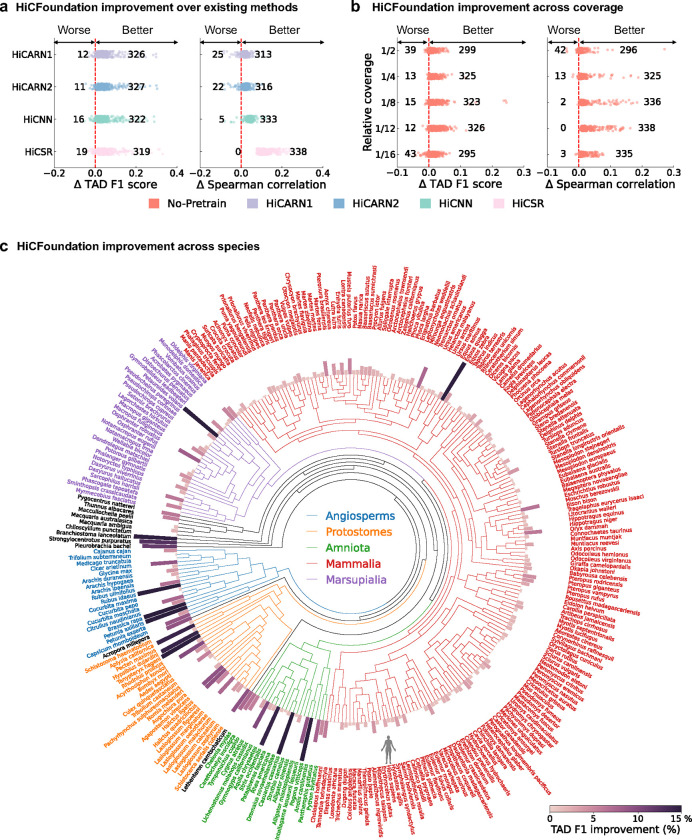
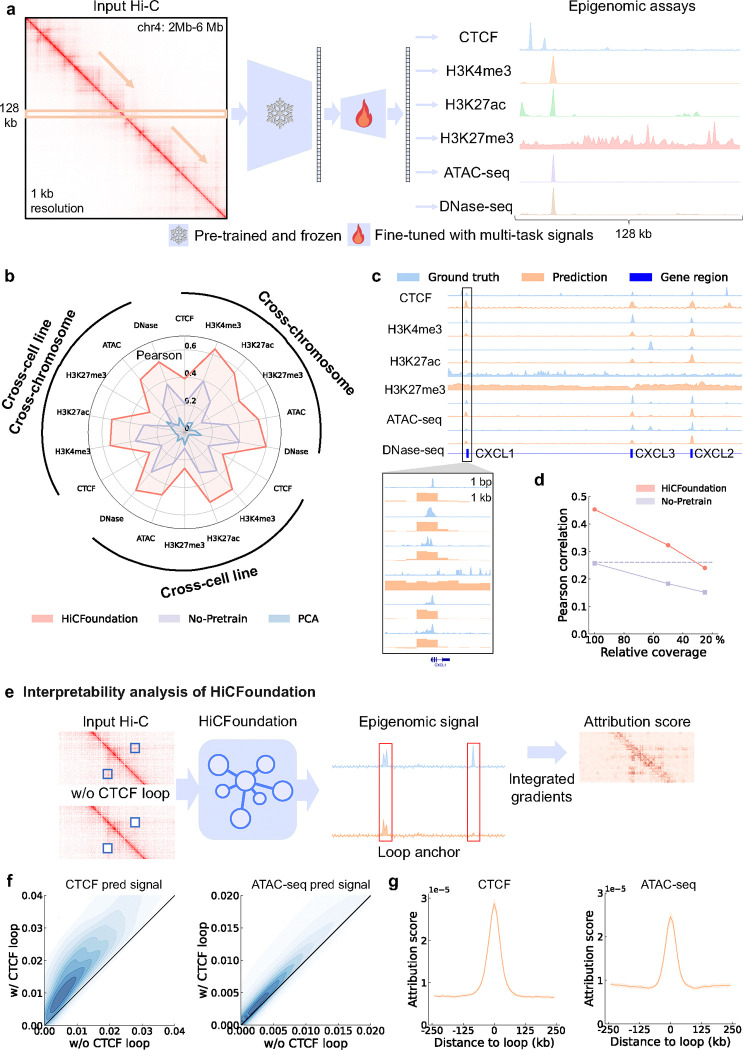
Nuclear DNA is organized into a compact three-dimensional (3D) structure that impacts critical cellular processes. High-throughput chromosome conformation capture (Hi-C) is the most widely used method for measuring 3D genome architecture, while linear epigenomic assays, such as ATAC-seq, DNase-seq, and ChIP-seq, are extensively employed to characterize epigenomic regulation. However, the integrative analysis of chromatin interactions and associated epigenomic regulation remains challenging due to the pairwise nature of Hi-C data, mismatched resolution between Hi-C and epigenomic assays, and inconsistencies among analysis tools. Here we propose HiCFoundation, a Hi-C-based foundation model for integrative analysis linking chromatin structure to downstream regulatory function. HiCFoundation is trained from hundreds of Hi-C assays encompassing 118 million contact matrix submatrices. The model achieves state-of-the-art performance on multiple types of 3D genome analysis, including reproducibility analysis, resolution enhancement, and loop detection. We further demonstrate the model's generalizability through genome architecture analysis of 316 species. Notably, by enhancing low-coverage experimental Hi-C data, HiCFoundation reveals genome-wide loop loss during differentiation of hematopoietic stem and progenitor cells (HSPCs) to neutrophils. Additionally, HiCFoundation is able to predict multiple types of epigenomic activity from Hi-C input and further interprets the link between Hi-C input and epigenomic output to reveal the relationship between chromatin conformation and genome function. Finally, HiCFoundation can analyze single-cell Hi-C data, shedding light on genome structure at single-cell resolution. HiCFoundation thus provides a unified, efficient, generalizable, and interpretable foundation for genome architecture, single-cell and multi-omics analysis across species, paving the path for systematically studying genome 3D architecture and its regulatory mechanisms.
核DNA被组织成一种紧凑的三维(3D)结构,这种结构会影响关键的细胞过程。高通量染色体构象捕获(Hi-C)是测量3D基因组结构最广泛使用的方法,而线性表观基因组分析,如ATAC-seq、DNase-seq和ChIP-seq,则被广泛用于表征表观基因组调控。然而,由于Hi-C数据的成对性质、Hi-C与表观基因组分析之间不匹配的分辨率以及分析工具之间的不一致性,染色质相互作用与相关表观基因组调控的综合分析仍然具有挑战性。在这里,我们提出了HiCFoundation,这是一种基于Hi-C的基础模型,用于将染色质结构与下游调控功能联系起来的综合分析。HiCFoundation是从数百个Hi-C实验中训练出来的,这些实验包含1.18亿个接触矩阵子矩阵。该模型在多种类型的3D基因组分析中取得了领先的性能,包括可重复性分析、分辨率增强和环检测。我们通过对316个物种的基因组结构分析进一步证明了该模型的通用性。值得注意的是,通过增强低覆盖度的实验性Hi-C数据,HiCFoundation揭示了造血干细胞和祖细胞(HSPCs)向中性粒细胞分化过程中全基因组范围的环丢失。此外,HiCFoundation能够从Hi-C输入预测多种类型的表观基因组活性,并进一步解释Hi-C输入与表观基因组输出之间的联系,以揭示染色质构象与基因组功能之间的关系。最后,HiCFoundation可以分析单细胞Hi-C数据,以单细胞分辨率揭示基因组结构。因此,HiCFoundation为跨物种的基因组结构、单细胞和多组学分析提供了一个统一、高效、通用且可解释的基础,为系统研究基因组3D结构及其调控机制铺平了道路。