Kim Jun, Park Jong Lyul, Yang Jin Ok, Kim Sangok, Joe Soobok, Park Gunwoo, Hwang Taeyeon, Cho Mun-Jeong, Lee Seungjae, Lee Jong-Eun, Park Ji-Hwan, Yeo Min-Kyung, Kim Seon-Young
Department of Convergent Bioscience and Informatics, College of Bioscience and Biotechnology, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea.
Personalized Genomic Medicine Research Center, Korea Research Institute of Bioscience & Biotechnology, 125, Gwahak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea.
Nucleic Acids Res. 2025 Jan 7;53(1). doi: 10.1093/nar/gkae1294.
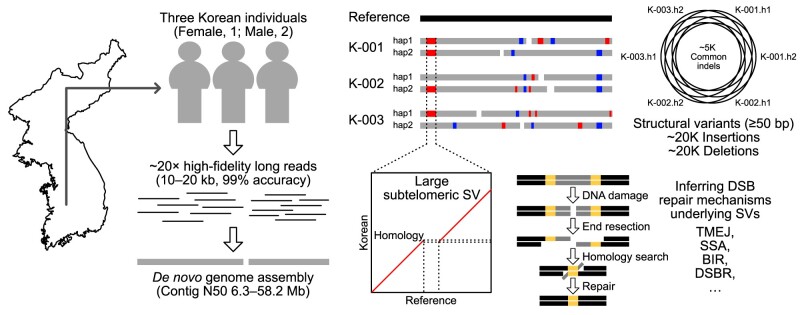
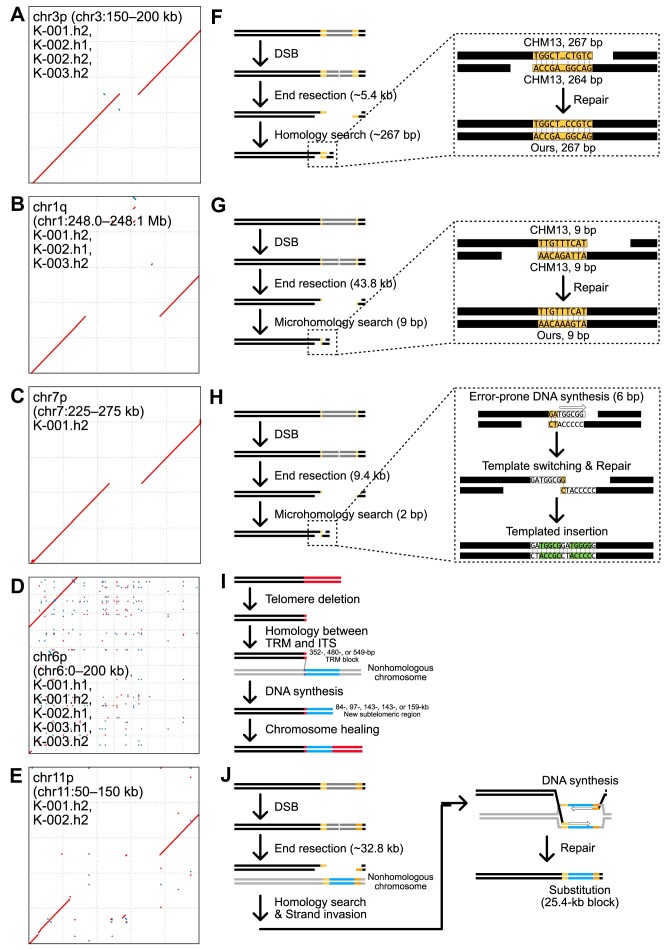
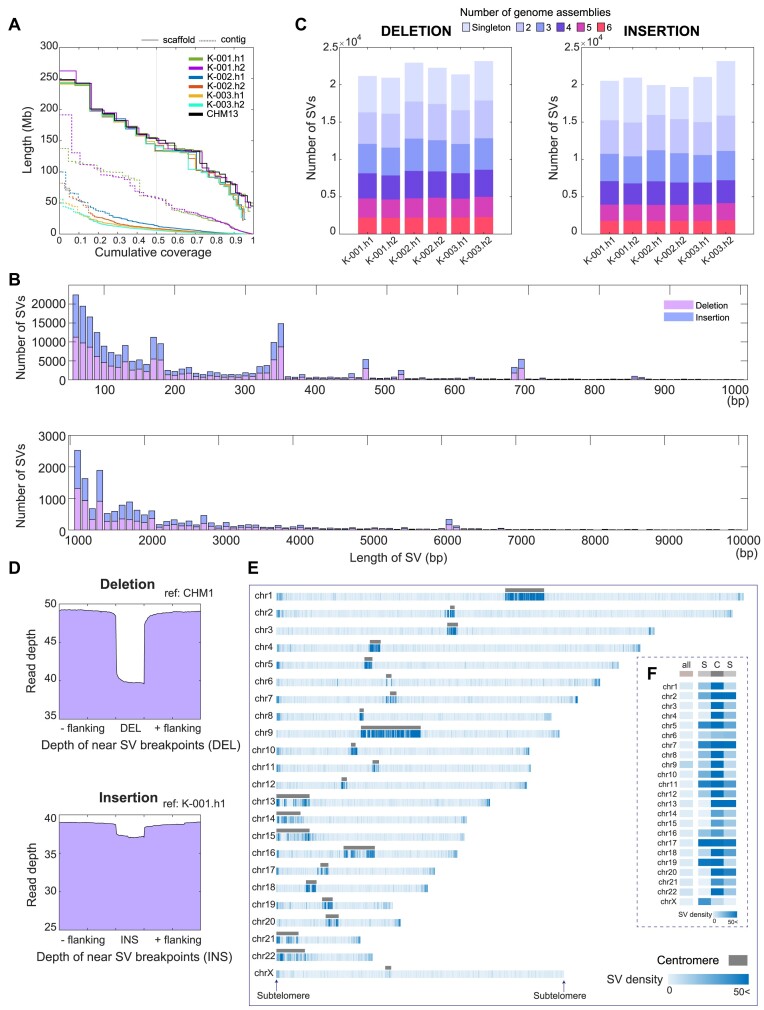
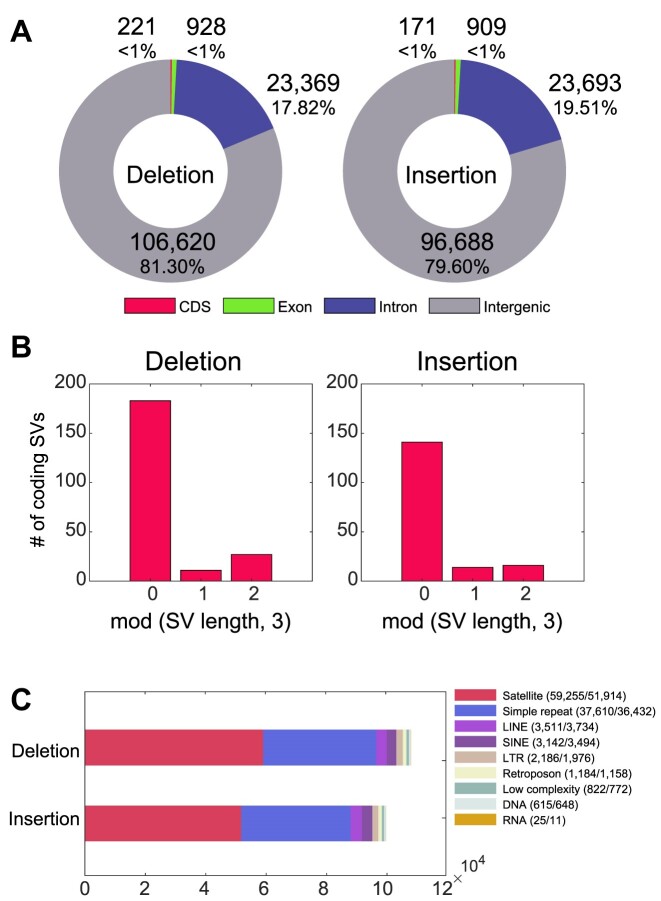
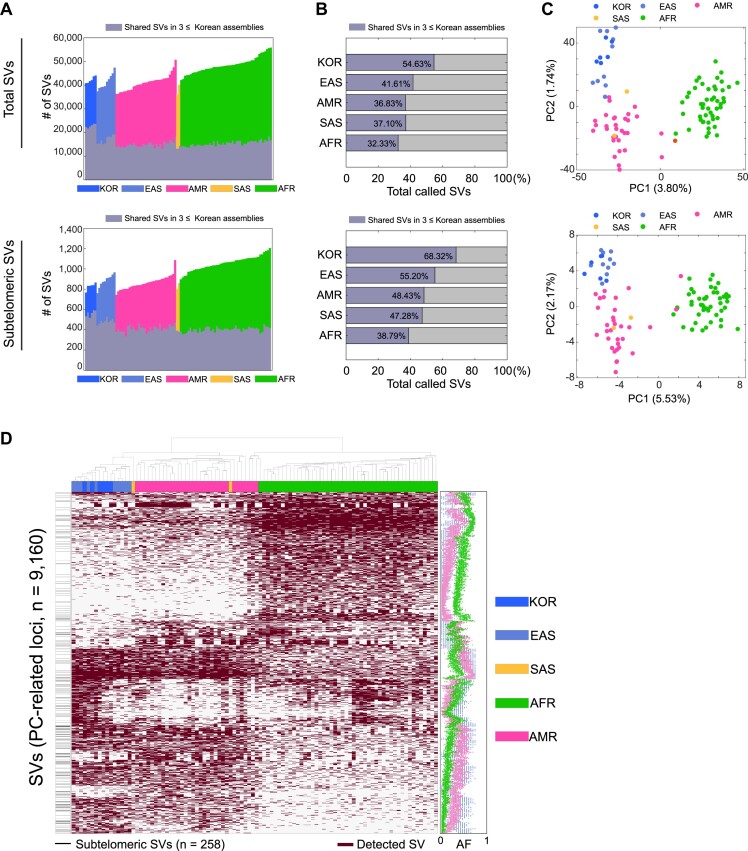
Given the presence of highly repetitive genomic regions such as subtelomeric regions, understanding human genomic evolution remains challenging. Recently, long-read sequencing technology has facilitated the identification of complex genetic variants, including structural variants (SVs), at the single-nucleotide level. Here, we resolved SVs and their underlying DNA damage-repair mechanisms in subtelomeric regions, which are among the most uncharted genomic regions. We generated ∼20 × high-fidelity long-read sequencing data from three Korean individuals and their partially phased high-quality de novo genome assemblies (contig N50: 6.3-58.2 Mb). We identified 131 138 deletion and 121 461 insertion SVs, 41.6% of which were prevalent in the East Asian population. The commonality of the SVs identified among the Korean population was examined by short-read sequencing data from 103 Korean individuals, providing the first comprehensive SV set representing the population based on the long-read assemblies. Manual investigation of 19 large subtelomeric SVs (≥5 kb) and their associated repair signatures revealed the potential repair mechanisms leading to the formation of these SVs. Our study provides mechanistic insight into human telomere evolution and can facilitate our understanding of human SV formation.
鉴于存在高度重复的基因组区域,如亚端粒区域,理解人类基因组进化仍然具有挑战性。最近,长读长测序技术有助于在单核苷酸水平上识别复杂的遗传变异,包括结构变异(SVs)。在这里,我们解析了亚端粒区域中的结构变异及其潜在的DNA损伤修复机制,亚端粒区域是最未知的基因组区域之一。我们从三名韩国个体及其部分定相的高质量从头基因组组装(重叠群N50:6.3-58.2 Mb)中生成了约20倍的高保真长读长测序数据。我们鉴定出131138个缺失和121461个插入结构变异,其中41.6%在东亚人群中普遍存在。通过对103名韩国个体的短读长测序数据检查韩国人群中鉴定出的结构变异的共性,基于长读长组装提供了代表该人群的首个全面的结构变异集。对19个大的亚端粒结构变异(≥5 kb)及其相关修复特征的人工研究揭示了导致这些结构变异形成的潜在修复机制。我们的研究为人类端粒进化提供了机制性见解,并有助于我们理解人类结构变异的形成。