Ishigohoka Jun, Liedvogel Miriam
Max Planck Research Group Behavioural Genomics, Max Planck Institute for Evolutionary Biology, August-Thienemann-Straße 2, Plön 24306, Germany.
Institute of Avian Research, An der Vogelwarte 21, Wilhelmshaven 26386, Germany.
Genetics. 2025 Mar 17;229(3). doi: 10.1093/genetics/iyaf004.
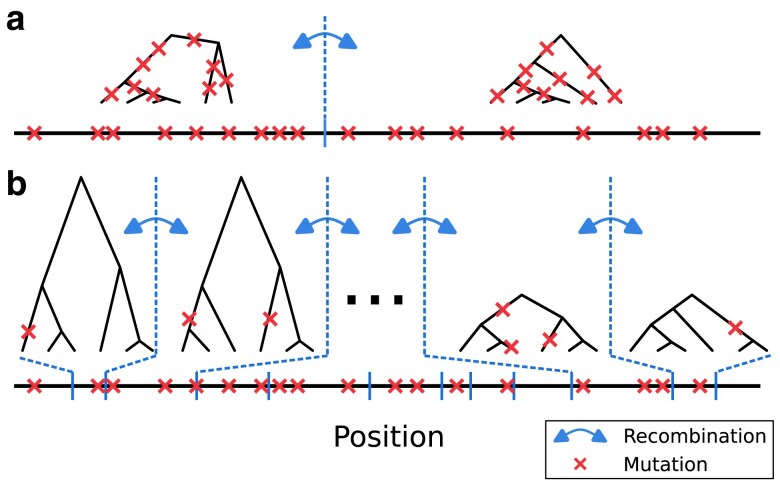
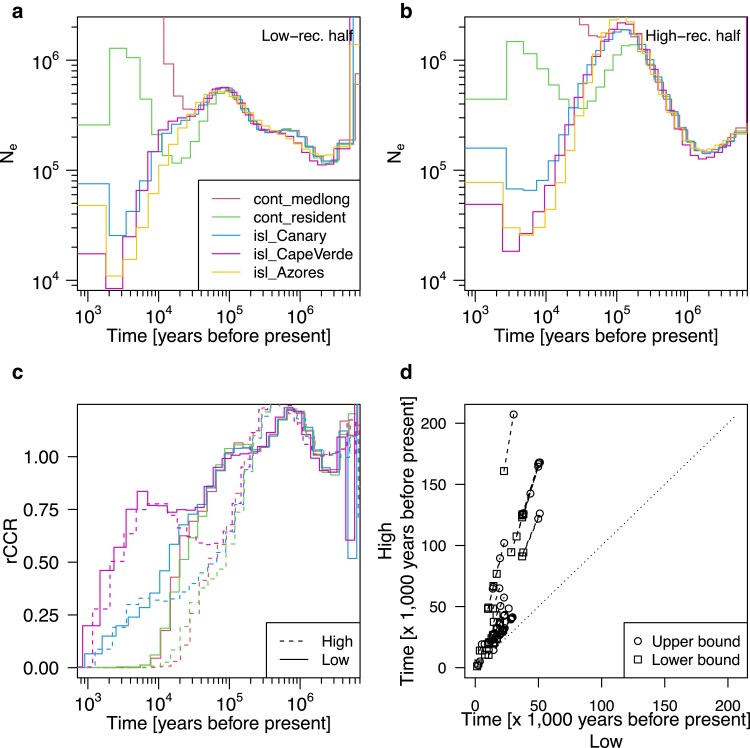
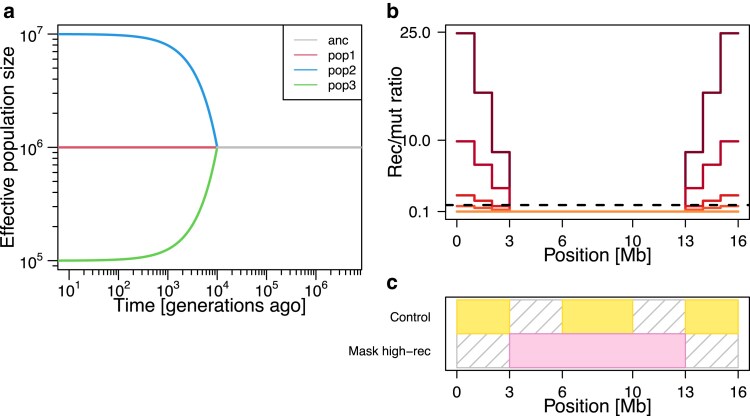
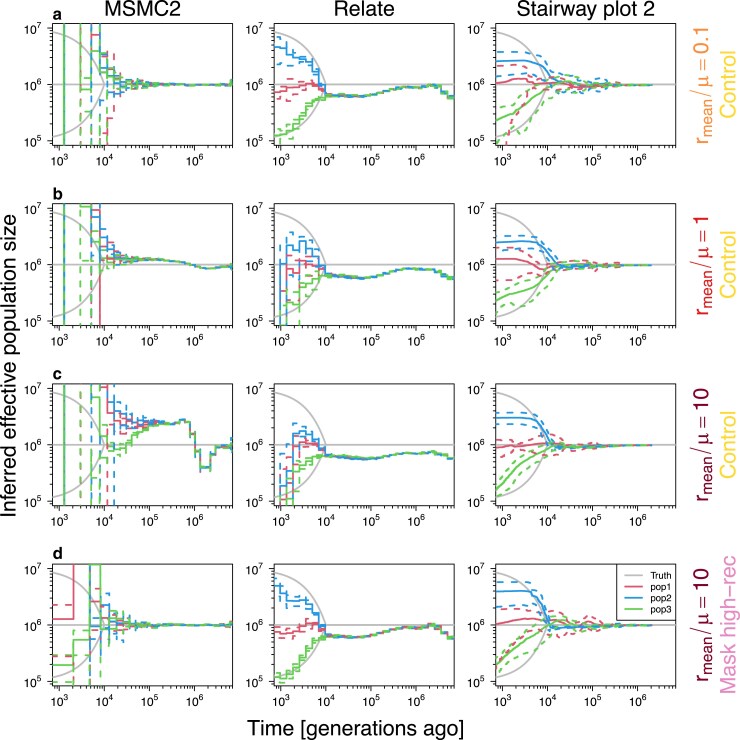
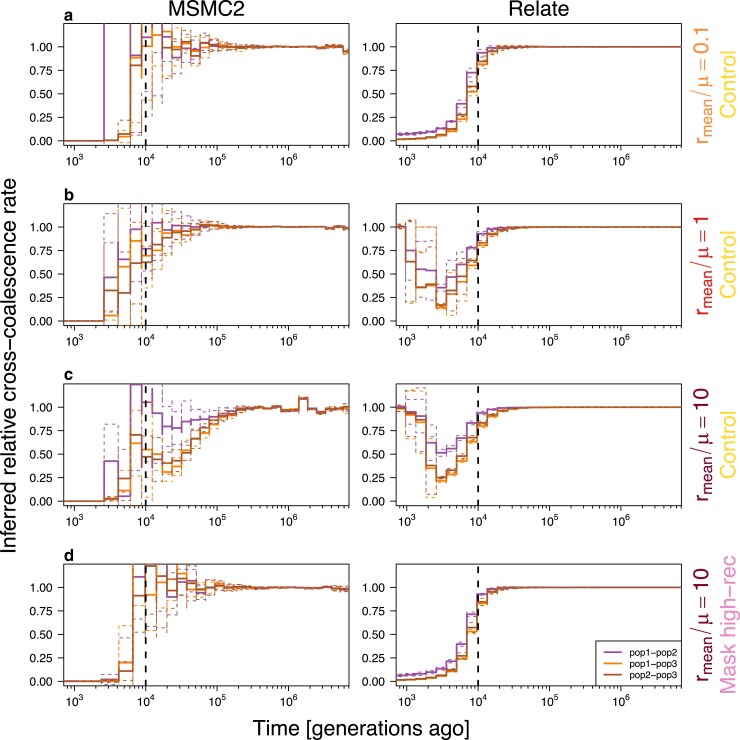
Multiple methods of demography inference are based on the ancestral recombination graph. This powerful approach uses observed mutations to model local genealogies changing along chromosomes by historical recombination events. However, inference of underlying genealogies is difficult in regions with high recombination rate relative to mutation rate due to the lack of mutations representing genealogies. Despite the prevalence of high-recombining genomic regions in some organisms, such as birds, its impact on demography inference based on ancestral recombination graphs has not been well studied. Here, we use population genomic simulations to investigate the impact of high-recombining regions on demography inference based on ancestral recombination graphs. We demonstrate that inference of effective population size and the time of population split events is systematically affected when high-recombining regions cover wide breadths of the chromosomes. Excluding high-recombining genomic regions can practically mitigate this impact, and population genomic inference of recombination maps is informative in defining such regions although the estimated values of local recombination rate can be biased. Finally, we confirm the relevance of our findings in empirical analysis by contrasting demography inferences applied for a bird species, the Eurasian blackcap (Sylvia atricapilla), using different parts of the genome with high and low recombination rates. Our results suggest that demography inference methods based on ancestral recombination graphs should be carried out with caution when applied in species whose genomes contain long stretches of high-recombining regions.
多种人口统计学推断方法基于祖先重组图。这种强大的方法利用观察到的突变来模拟由于历史重组事件而沿染色体变化的局部谱系。然而,在重组率相对于突变率较高的区域,由于缺乏代表谱系的突变,推断潜在的谱系是困难的。尽管在一些生物(如鸟类)中高重组基因组区域普遍存在,但它对基于祖先重组图的人口统计学推断的影响尚未得到充分研究。在这里,我们使用群体基因组模拟来研究高重组区域对基于祖先重组图的人口统计学推断的影响。我们证明,当高重组区域覆盖染色体的较宽范围时,有效种群大小和种群分裂事件时间的推断会受到系统性影响。排除高重组基因组区域实际上可以减轻这种影响,并且重组图谱的群体基因组推断在定义此类区域方面具有参考价值,尽管局部重组率的估计值可能存在偏差。最后,我们通过对比对一种鸟类——欧亚褐头山雀(Sylvia atricapilla)应用不同重组率高低的基因组部分进行的人口统计学推断,在实证分析中证实了我们研究结果的相关性。我们的结果表明,基于祖先重组图的人口统计学推断方法在应用于基因组包含长片段高重组区域的物种时应谨慎使用。