Kankipati Sri Meghana, Dumra Surbhi, Thareja Swati, Ishfaq Lyluma, Zargar Mah N, Das Arghadip, Kongala Sreya, Younas Salma
Medicine and Surgery, Andhra Medical College, Visakhapatnam, IND.
Medicine, Employees' State Insurance Corporation (ESIC) Medical College and Hospital, Faridabad, IND.
Cureus. 2024 Dec 9;16(12):e75434. doi: 10.7759/cureus.75434. eCollection 2024 Dec.
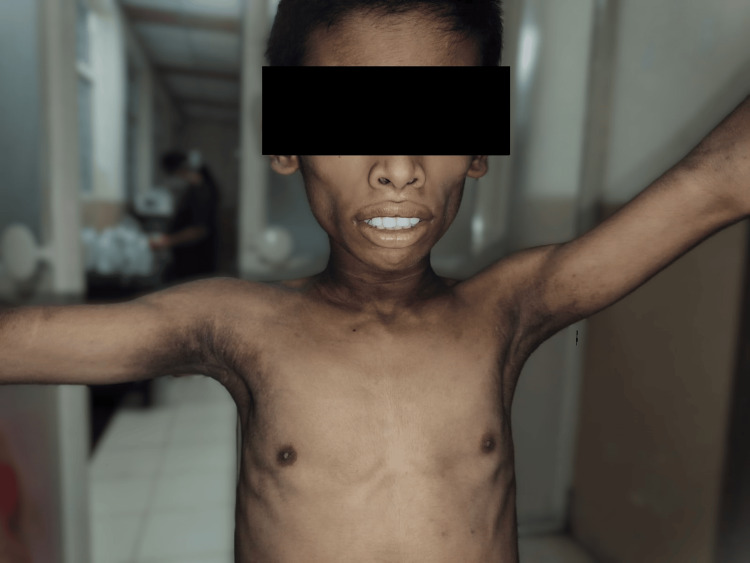
Berardinelli-Seip congenital lipodystrophy (BSCL), also known as congenital generalized lipodystrophy (CGL), is an exceptionally rare autosomal recessive disorder marked by a significant deficiency of adipose tissue throughout the body. This lack of adipose tissue, normally found beneath the skin and between internal organs, leads to impaired adipocyte formation and fat storage, causing lipids to accumulate in atypical tissues such as muscles and the liver. The extent of adipose tissue loss directly influences the severity of symptoms, which can include a muscular appearance, increased appetite, bone cysts, marrow fat depletion, acromegalic features, severe insulin resistance, skeletal muscle hypertrophy, hypertrophic cardiomyopathy, hepatic steatosis, hepatomegaly, cirrhosis, and intellectual disability. We present a case series of two siblings with BSCL: a nine-year-old boy and his seven-year-old sister, each with unique manifestations of the disorder. The older sibling presented with high-grade fever and right ankle pain, possibly indicative of a calcified deposit, alongside complications such as hyperglycemia (managed without insulin) and moderate pulmonary arterial hypertension (PAH) with tricuspid regurgitation (TR). The younger sibling displayed similar metabolic and cardiovascular complications, including hepatomegaly and early signs of cardiac involvement. Both cases required comprehensive evaluations, revealing anemia, thrombocytopenia, elevated leukocyte count, and high C-reactive protein (CRP) levels. The children were managed with high-potency antibiotics, leading to a marked improvement in their overall conditions. These cases demonstrate the broad spectrum of clinical manifestations associated with BSCL and highlight the importance of a multidisciplinary approach for effective management. Although limited by the small sample size, this case series shows the importance of a multidisciplinary approach in addressing the complex and overlapping symptoms of BSCL, which often mimic more common conditions. Increased awareness among healthcare providers is crucial for ensuring timely diagnosis and appropriate intervention, particularly in pediatric patients.
贝拉尔迪内利 - 塞普先天性脂肪营养不良(BSCL),也称为先天性全身性脂肪营养不良(CGL),是一种极为罕见的常染色体隐性疾病,其特征是全身脂肪组织严重缺乏。这种通常存在于皮肤下方和内脏器官之间的脂肪组织缺失,导致脂肪细胞形成和脂肪储存受损,使脂质在非典型组织如肌肉和肝脏中积聚。脂肪组织缺失的程度直接影响症状的严重程度,症状可能包括肌肉外观、食欲增加、骨囊肿、骨髓脂肪消耗、肢端肥大症特征、严重胰岛素抵抗、骨骼肌肥大、肥厚型心肌病、肝脂肪变性、肝肿大、肝硬化和智力残疾。我们报告了一例患有BSCL的两兄弟姐妹病例系列:一名9岁男孩和他7岁的妹妹,他们各自有该疾病的独特表现。年长一点的兄弟姐妹出现高热和右踝疼痛,可能提示有钙化沉积物,同时伴有高血糖(无需胰岛素治疗)和中度肺动脉高压(PAH)伴三尖瓣反流(TR)等并发症。年幼一点的兄弟姐妹表现出类似的代谢和心血管并发症,包括肝肿大和心脏受累的早期迹象。两例均需要全面评估,结果显示有贫血、血小板减少、白细胞计数升高和高C反应蛋白(CRP)水平。这两个孩子接受了强效抗生素治疗,整体状况有显著改善。这些病例证明了与BSCL相关的广泛临床表现,并强调了多学科方法对有效管理的重要性。尽管受样本量小的限制,但这个病例系列显示了多学科方法在解决BSCL复杂且重叠的症状(这些症状常常类似更常见的病症)方面的重要性。提高医疗服务提供者的认识对于确保及时诊断和适当干预至关重要,尤其是在儿科患者中。