Division of Pediatrics, Department of Health Sciences, University of Piemonte Orientale, Novara, Italy.
Endocrinology, Department of Translational Medicine, University of Piemonte Orientale, Novara, Italy.
Front Endocrinol (Lausanne). 2023 Jul 12;14:1212729. doi: 10.3389/fendo.2023.1212729. eCollection 2023.
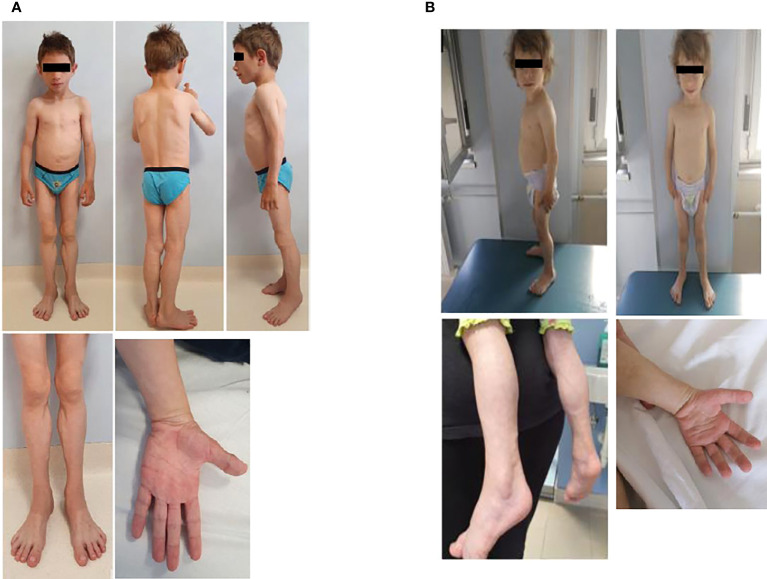
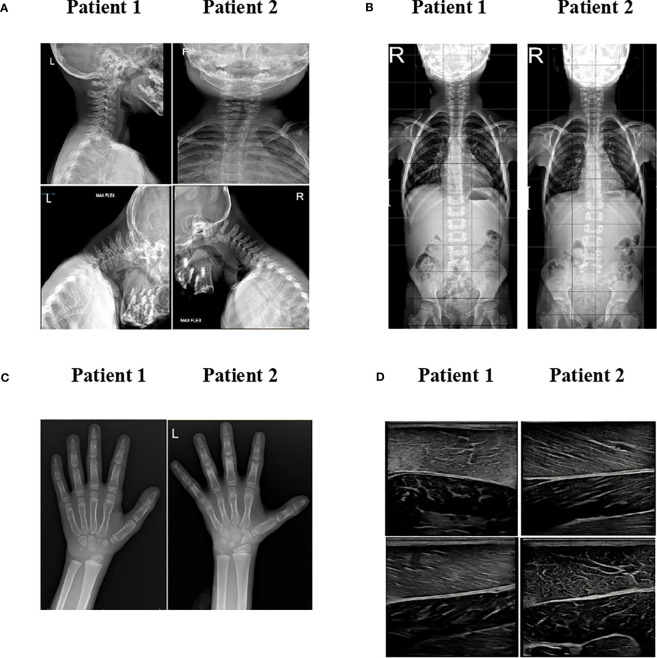
Lipodystrophy syndromes are characterized by a progressive metabolic impairment secondary to adipose tissue dysfunction and may have a genetic background. Congenital generalized lipodystrophy type 4 (CGL4) is an extremely rare subtype, caused by mutations in the polymerase I and transcript release factor () gene. It encodes for a cytoplasmatic protein called caveolae-associated protein 1 (Cavin-1), which, together with caveolin 1, is responsible for the biogenesis of caveolae, being a master regulator of adipose tissue expandability. Cavin-1 is expressed in several tissues, including muscles, thus resulting, when dysfunctional, in a clinical phenotype characterized by the absence of adipose tissue and muscular dystrophy. We herein describe the clinical phenotypes of two siblings in their early childhood, with a phenotype characterized by a generalized reduction of subcutaneous fat, muscular hypertrophy, distinct facial features, myopathy, and atlantoaxial instability. One of the siblings developed paroxysmal supraventricular tachycardia leading to cardiac arrest at 3 months of age. Height and BMI were normal. Blood tests showed elevated CK, a mild increase in liver enzymes and triglycerides levels, and undetectable leptin and adiponectin concentrations. Fasting glucose and HbA1c were normal, while Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) was mildly elevated. Both patients were hyperphagic and had cravings for foods rich in fats and sugars. Genetic testing revealed a novel pathogenic mutation of the / gene (NM_012232 exon1:c T21A:p.Y7X) at the homozygous state. The diagnosis of lipodystrophy can be challenging, often requiring a multidisciplinary approach, given the pleiotropic effect, involving several tissues. The coexistence of generalized lack of fat, myopathy with elevated CK levels, arrhythmias, gastrointestinal dysmotility, and skeletal abnormalities should prompt the suspicion for the diagnosis of CGL4, although phenotypic variability may occur.
脂肪营养不良综合征的特征是脂肪组织功能障碍导致的进行性代谢损伤,可能具有遗传背景。先天性全身性脂肪营养不良 4 型(CGL4)是一种极其罕见的亚型,由聚合酶 I 和转录释放因子()基因突变引起。它编码一种细胞质蛋白,称为小窝相关蛋白 1(Cavin-1),它与 caveolin 1 一起负责小窝的生物发生,是脂肪组织可扩展性的主要调节因子。Cavin-1 在包括肌肉在内的多种组织中表达,因此当其功能失调时,会导致以缺乏脂肪组织和肌肉营养不良为特征的临床表现型。我们在此描述了两名在幼儿期的同胞的临床表现型,其表现型特征为皮下脂肪普遍减少、肌肉肥大、独特的面部特征、肌病和寰枢椎不稳定。其中一名同胞在 3 个月大时发生阵发性室上性心动过速导致心脏骤停。身高和 BMI 正常。血液检查显示 CK 升高,肝酶和甘油三酯水平轻度升高,瘦素和脂联素浓度无法检测到。空腹血糖和 HbA1c 正常,而胰岛素抵抗稳态模型评估(HOMA-IR)轻度升高。两名患者均食欲旺盛,渴望富含脂肪和糖的食物。基因检测显示 / 基因(NM_012232 外显子 1:c T21A:p.Y7X)的新型致病性突变呈纯合状态。由于涉及多个组织的多效性效应,脂肪营养不良的诊断具有挑战性,通常需要多学科方法。广泛性脂肪缺乏、肌病伴 CK 水平升高、心律失常、胃肠道动力障碍和骨骼异常的共存应提示怀疑 CGL4 的诊断,尽管可能存在表型变异性。