Jiang Yafeng, Hu Zhaolan, Huang Roujie, Ho Kaying, Wang Pengfei, Kang Jin
Department of Hematology, the Second Xiangya Hospital of Central South University, Changsha, China.
Department of Anesthesiology, The Second Xiangya Hospital, Central South University, Changsha, China.
Front Immunol. 2025 Jan 3;15:1512483. doi: 10.3389/fimmu.2024.1512483. eCollection 2024.
Anti-citrullinated peptide antibodies (ACPA)-negative (ACPA-) rheumatoid arthritis (RA) presents significant diagnostic and therapeutic challenges due to the absence of specific biomarkers, underscoring the need to elucidate its distinctive cellular and metabolic profiles for more targeted interventions.
Single-cell RNA sequencing data from peripheral blood mononuclear cells (PBMCs) and synovial tissues of patients with ACPA- and ACPA+ RA, as well as healthy controls, were analyzed. Immune cell populations were classified based on clustering and marker gene expression, with pseudotime trajectory analysis, weighted gene co-expression network analysis (WGCNA), and transcription factor network inference providing further insights. Cell-cell communication was explored using CellChat and MEBOCOST, while scFEA enabled metabolic flux estimation. A neural network model incorporating key genes was constructed to differentiate patients with ACPA- RA from healthy controls.
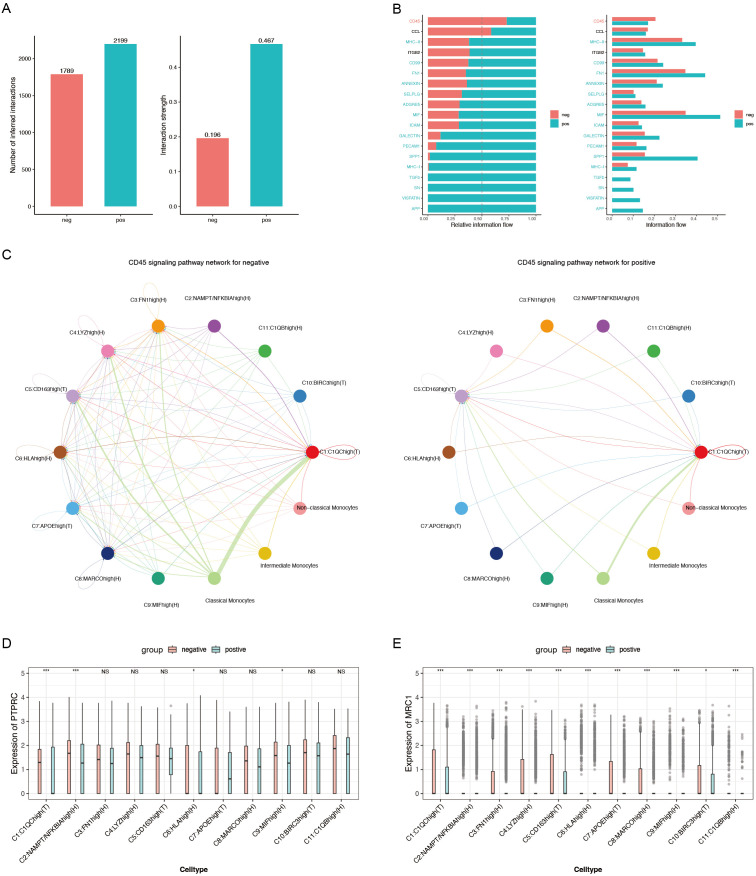
Patients with ACPA- RA demonstrated a pronounced increase in classical monocytes in PBMCs and C1QChigh macrophages (p < 0.001 and p < 0.05). Synovial macrophages exhibited increased heterogeneity and were enriched in distinct metabolic pathways, including complement cascades and glutathione metabolism. The neural network model achieved reliable differentiation between patients with ACPA- RA and healthy controls (AUC = 0.81). CellChat analysis identified CD45 and CCL5 as key pathways facilitating macrophage-monocyte interactions in ACPA- RA, prominently involving iron-mediated metabolite communication. Metabolic flux analysis indicated elevated beta-alanine and glutathione metabolism in ACPA- RA macrophages.
These findings underscore that ACPA-negative rheumatoid arthritis is marked by elevated classical monocytes in circulation and metabolic reprogramming of synovial macrophages, particularly in complement cascade and glutathione metabolism pathways. By integrating single-cell RNA sequencing with machine learning, this study established a neural network model that robustly differentiates patients with ACPA- RA from healthy controls, highlighting promising diagnostic biomarkers and therapeutic targets centered on immune cell metabolism.
抗瓜氨酸化肽抗体(ACPA)阴性(ACPA-)类风湿性关节炎(RA)由于缺乏特异性生物标志物,在诊断和治疗上面临重大挑战,这突出表明需要阐明其独特的细胞和代谢特征,以便进行更有针对性的干预。
分析了ACPA-和ACPA+ RA患者以及健康对照者外周血单核细胞(PBMC)和滑膜组织的单细胞RNA测序数据。基于聚类和标记基因表达对免疫细胞群体进行分类,通过伪时间轨迹分析、加权基因共表达网络分析(WGCNA)和转录因子网络推断进一步深入了解。使用CellChat和MEBOCOST探索细胞间通讯,而scFEA可进行代谢通量估计。构建了一个包含关键基因的神经网络模型,以区分ACPA- RA患者和健康对照者。
ACPA- RA患者的PBMC中经典单核细胞以及C1QChigh巨噬细胞显著增加(p < 0.001和p < 0.05)。滑膜巨噬细胞表现出更高的异质性,并富集于不同的代谢途径,包括补体级联反应和谷胱甘肽代谢。神经网络模型在ACPA- RA患者和健康对照者之间实现了可靠的区分(AUC = 0.81)。CellChat分析确定CD45和CCL5是促进ACPA- RA中巨噬细胞 - 单核细胞相互作用的关键途径,主要涉及铁介导的代谢物通讯。代谢通量分析表明ACPA- RA巨噬细胞中β-丙氨酸和谷胱甘肽代谢升高。
这些发现强调,ACPA阴性类风湿性关节炎的特征是循环中经典单核细胞升高以及滑膜巨噬细胞的代谢重编程,特别是在补体级联反应和谷胱甘肽代谢途径中。通过将单细胞RNA测序与机器学习相结合,本研究建立了一个能够可靠区分ACPA- RA患者和健康对照者的神经网络模型,突出了以免疫细胞代谢为中心的有前景的诊断生物标志物和治疗靶点。