Almuhaideb Esam, Hasan Nur A, Grim Christopher, Rashed Shah Manzur, Parveen Salina
Department of Agriculture, Food and Resource Sciences, University of Maryland Eastern Shore, Princess Anne, MD, United States.
Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, United States.
Front Microbiol. 2025 Jan 8;15:1504487. doi: 10.3389/fmicb.2024.1504487. eCollection 2024.
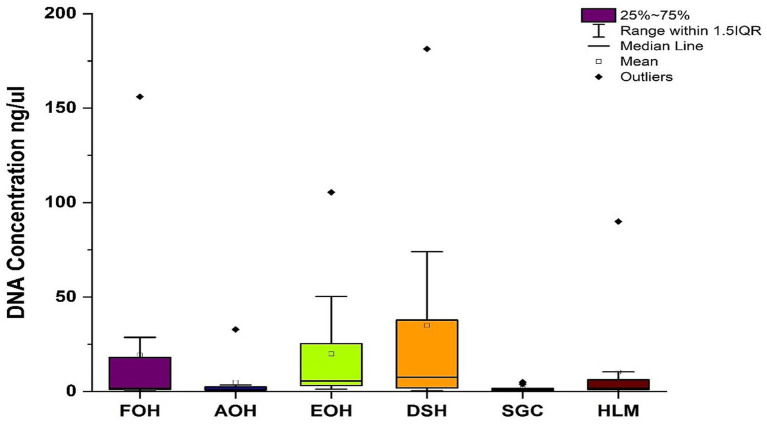
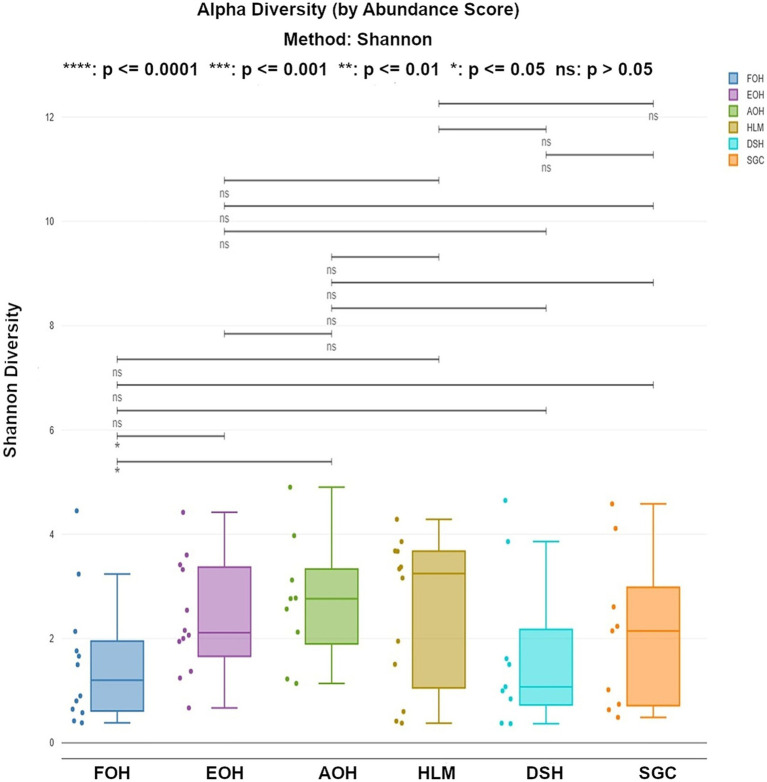
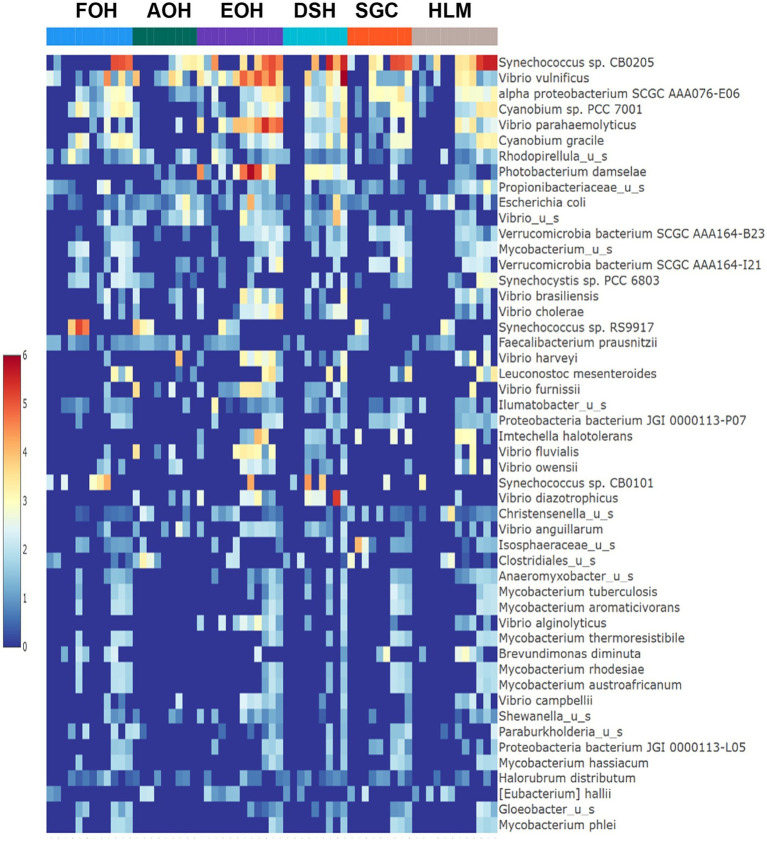
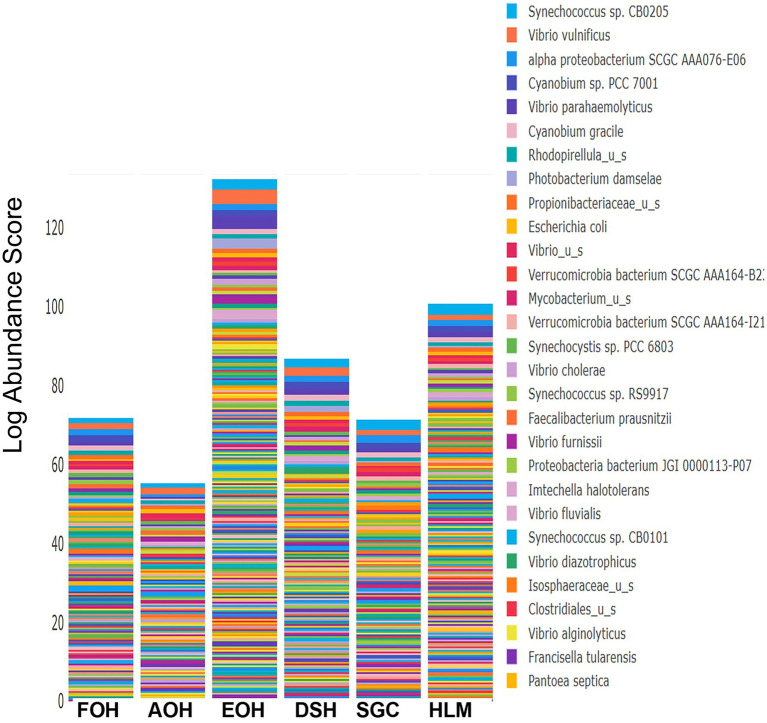
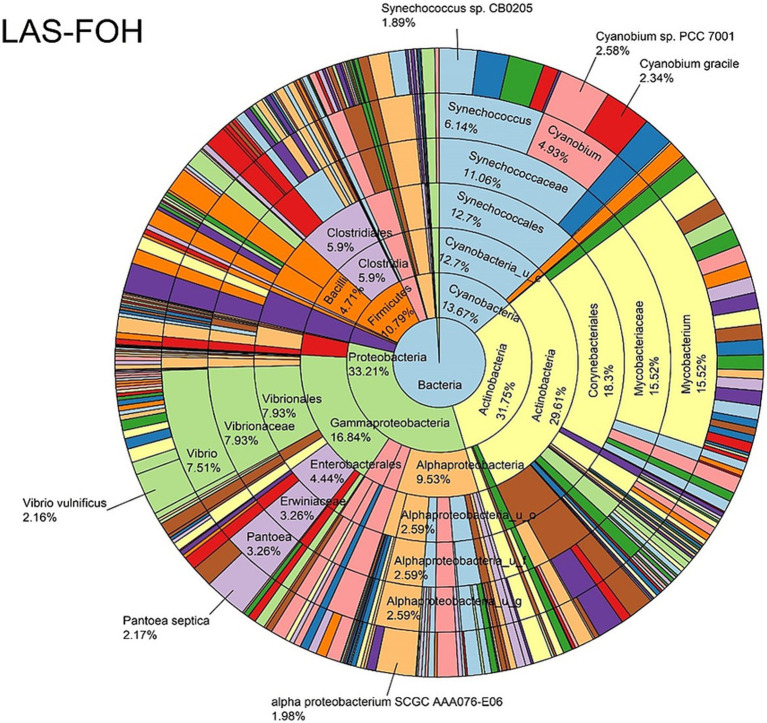
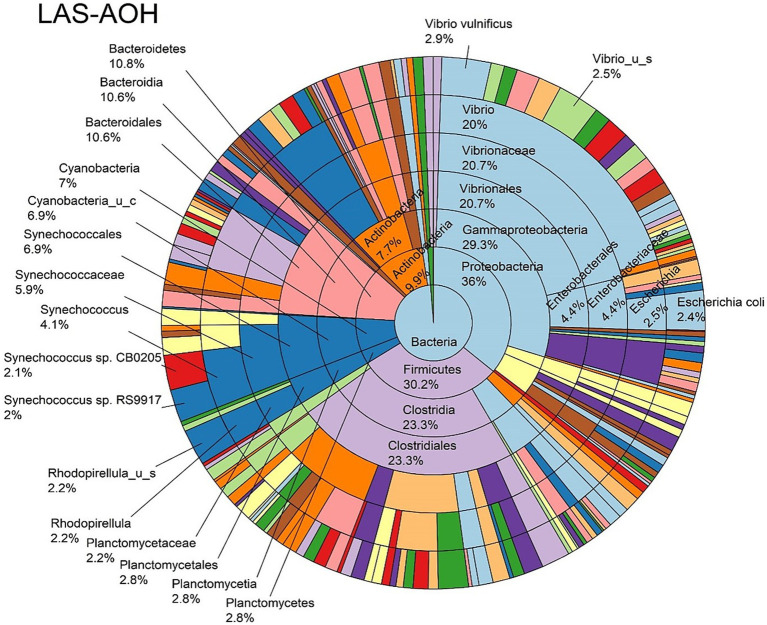
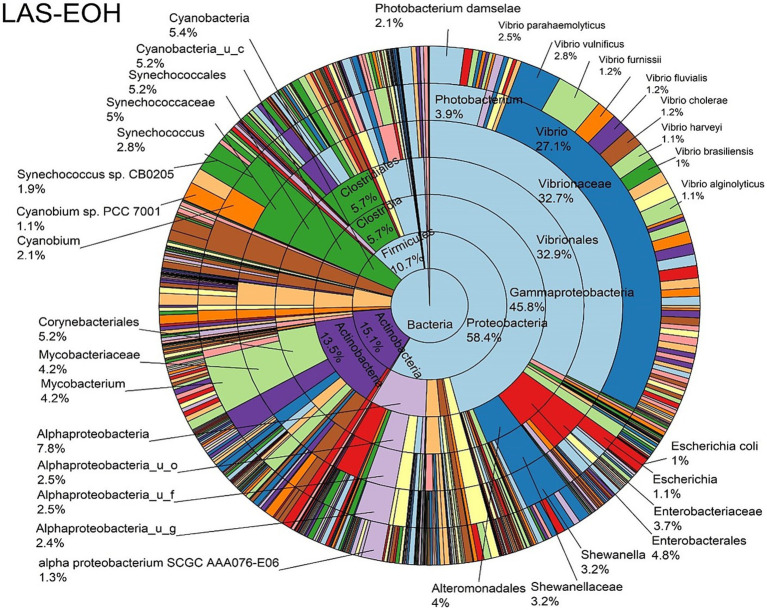
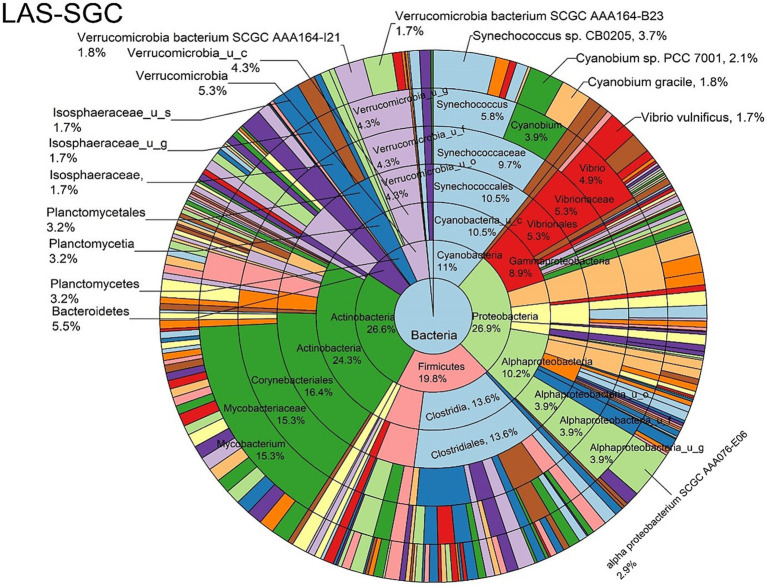
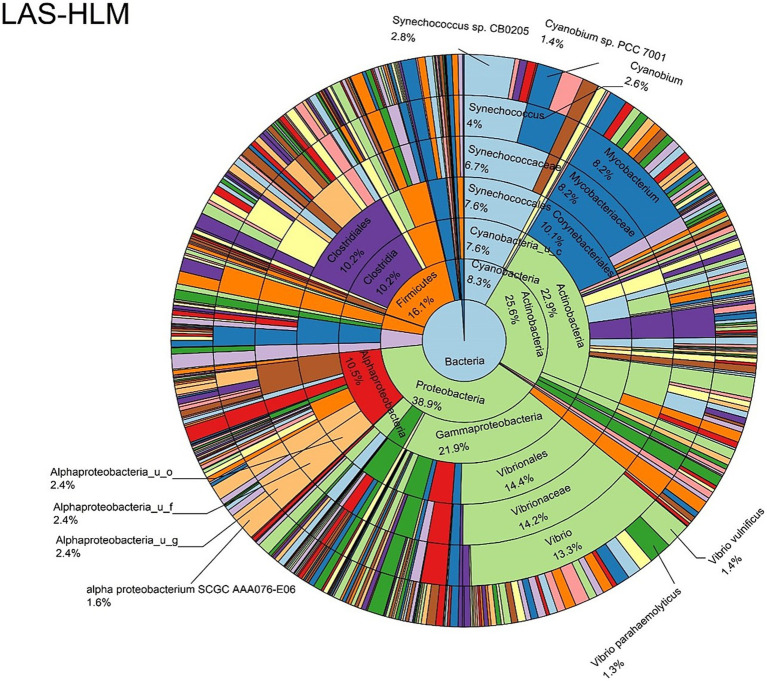
Metagenomic sequencing is increasingly being employed to understand the assemblage and dynamics of the oyster microbiome. Specimen collection and processing steps can impact the resultant microbiome composition and introduce bias. To investigate this systematically, a total of 54 farmed oysters were collected from Chesapeake Bay between May and September 2019. Six different specimen types and processing methods were evaluated for microbial community composition using shotgun metagenomics, namely fresh oyster homogenate (FOH), oyster homogenate after simulated temperature abuse (AOH), Luria broth-enriched oyster homogenate (EOH), dissected stomach homogenate (DSH), hemolymph (HLM), and stomach-gut content (SGC). In general, DSH, EOH, and FOH yielded the highest DNA concentration, while EOH had the highest microbial reads, followed by DSH, HLM, and FOH. HLM produced the highest bacterial species alpha diversity, followed by AOH, EOH, and SGC. Although alpha diversities did not differ significantly, beta-diversity measurements showed significant dissimilarity among methods ( < 0.05) indicating that the specimen types and processing steps do play an important role in representing the composition of the bacterial community. Bacterial species that had the highest log mean abundance included sp. PCC 7001 in FOH, in AOH, EOH, and DSH, and lastly sp. CB0205 in the DSH, HML, and SGC samples. EOH displayed higher bacterial hits, distinct microbial composition, and higher values of bacterial, phages, and antimicrobial resistance gene reads. Therefore, if studying the overall oyster microbial community, prioritizing optimum specimen collection and processing methods that align with the overall goal of the study is recommended.
宏基因组测序越来越多地被用于了解牡蛎微生物组的组成和动态。样本采集和处理步骤会影响最终的微生物组组成并引入偏差。为了系统地研究这一点,2019年5月至9月期间从切萨皮克湾总共采集了54只养殖牡蛎。使用鸟枪法宏基因组学评估了六种不同的样本类型和处理方法对微生物群落组成的影响,即新鲜牡蛎匀浆(FOH)、模拟温度滥用后的牡蛎匀浆(AOH)、富含Luria肉汤的牡蛎匀浆(EOH)、解剖后的胃匀浆(DSH)、血淋巴(HLM)和胃-肠内容物(SGC)。一般来说,DSH、EOH和FOH产生的DNA浓度最高,而EOH的微生物读数最高,其次是DSH、HLM和FOH。HLM产生的细菌物种α多样性最高,其次是AOH、EOH和SGC。虽然α多样性没有显著差异,但β多样性测量显示不同方法之间存在显著差异(<0.05),这表明样本类型和处理步骤在代表细菌群落组成方面确实起着重要作用。对数平均丰度最高的细菌物种包括FOH中的sp. PCC 7001,AOH、EOH和DSH中的 ,以及DSH、HML和SGC样本中的sp. CB0205。EOH显示出更高的细菌匹配度、独特的微生物组成以及细菌、噬菌体和抗菌抗性基因读数的更高值。因此,如果研究整个牡蛎微生物群落,建议优先选择与研究总体目标一致的最佳样本采集和处理方法。