Pena Izabella A, Shi Jeffrey S, Chang Sarah M, Yang Jason, Block Samuel, Adelmann Charles H, Keys Heather R, Ge Preston, Bathla Shveta, Witham Isabella H, Sienski Grzegorz, Nairn Angus C, Sabatini David M, Lewis Caroline A, Kory Nora, Vander Heiden Matthew G, Heiman Myriam
The Picower Institute for Learning and Memory, MIT, Cambridge, MA, USA.
Department of Brain and Cognitive Sciences, MIT, Cambridge, MA, USA.
Nat Commun. 2025 Jan 24;16(1):978. doi: 10.1038/s41467-025-56130-3.
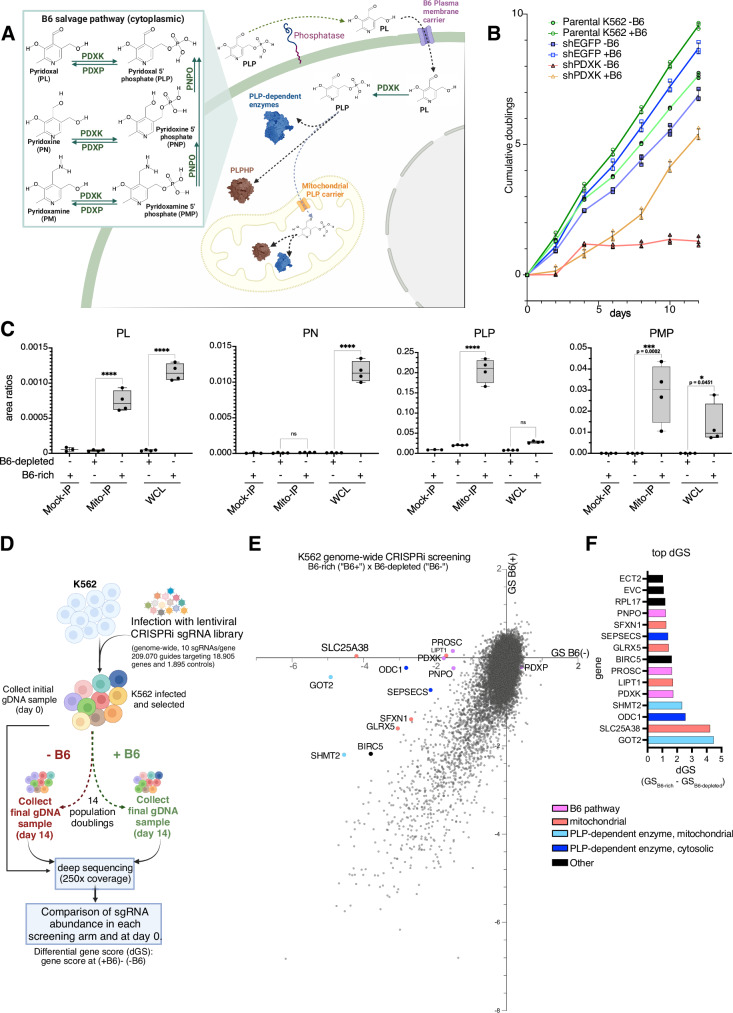
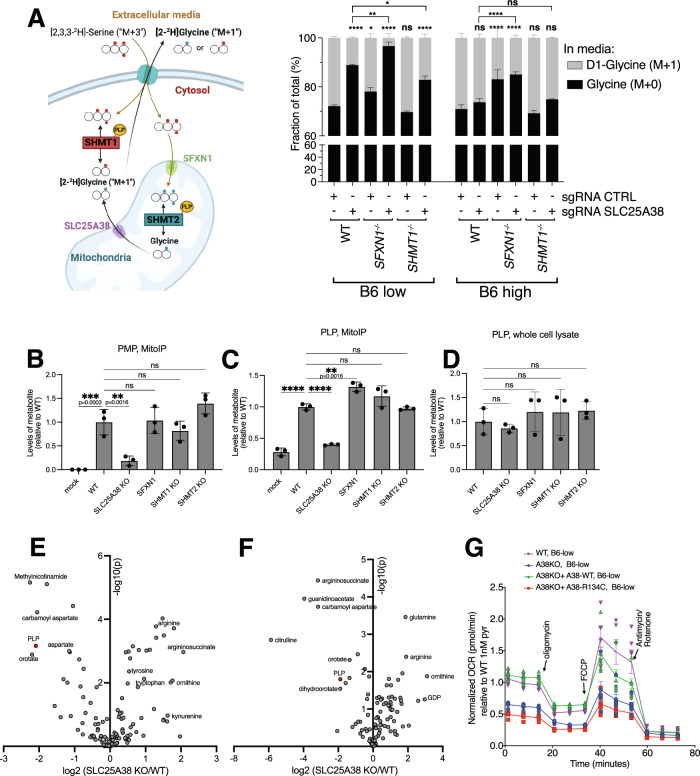
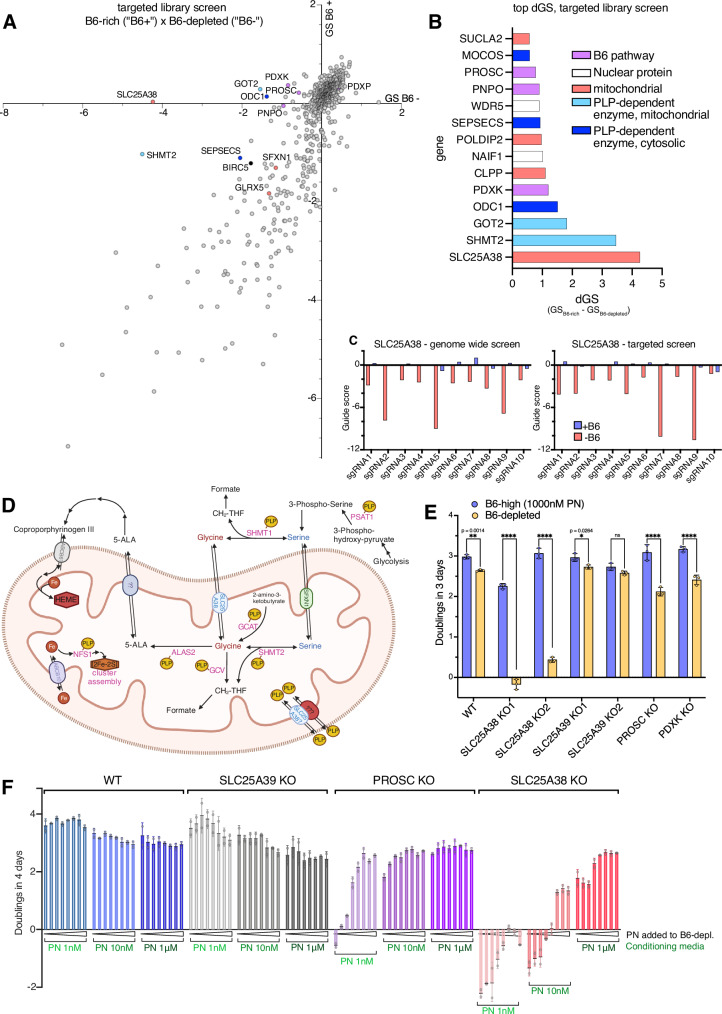
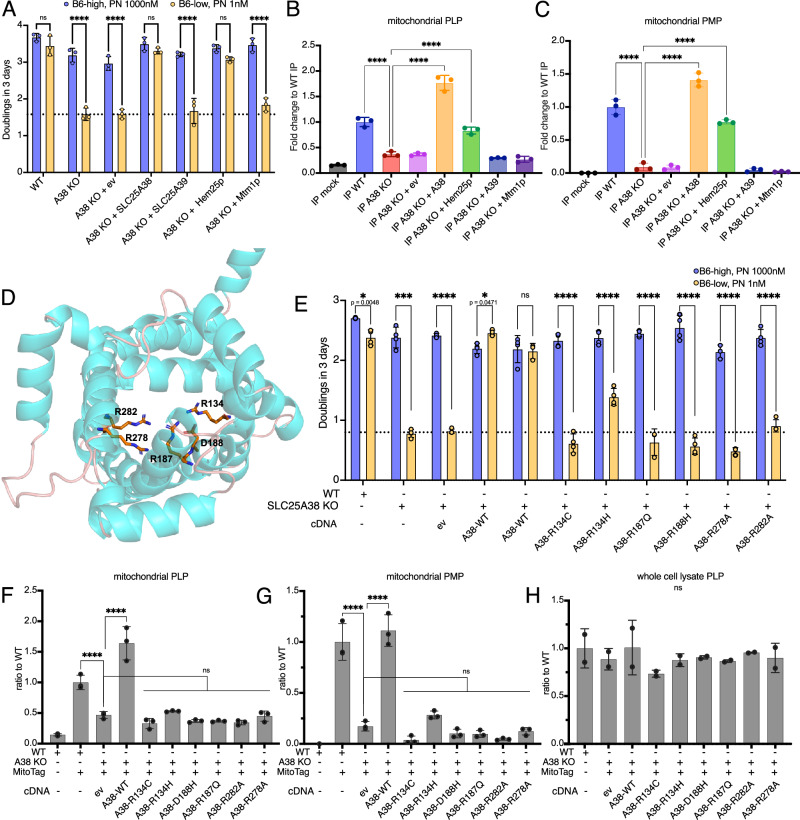
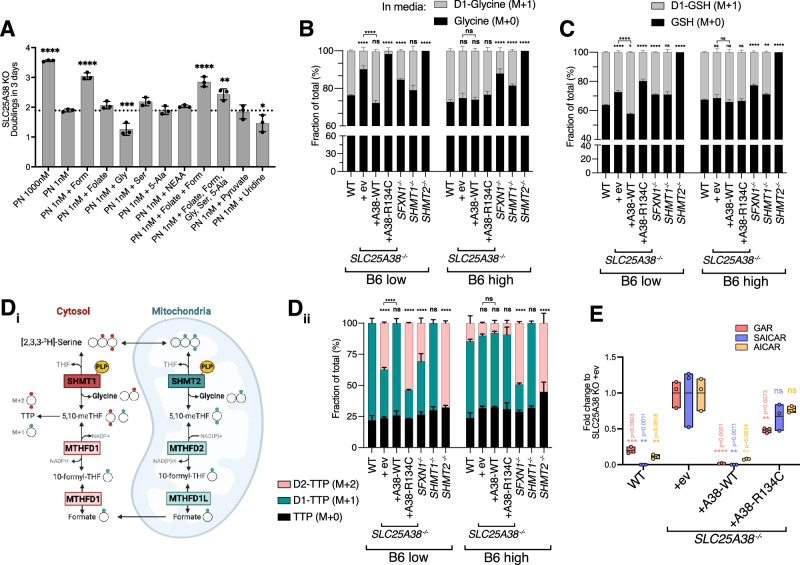
Many essential proteins require pyridoxal 5'-phosphate, the active form of vitamin B6, as a cofactor for their activity. These include enzymes important for amino acid metabolism, one-carbon metabolism, polyamine synthesis, erythropoiesis, and neurotransmitter metabolism. A third of all mammalian pyridoxal 5'-phosphate-dependent enzymes are localized in the mitochondria; however, the molecular machinery involved in the regulation of mitochondrial pyridoxal 5'-phosphate levels in mammals remains unknown. In this study, we used a genome-wide CRISPR interference screen in erythroleukemia cells and organellar metabolomics to identify the mitochondrial inner membrane protein SLC25A38 as a regulator of mitochondrial pyridoxal 5'-phosphate. Loss of SLC25A38 causes depletion of mitochondrial, but not cellular, pyridoxal 5'-phosphate, and impairs cellular proliferation under both physiological and low vitamin B6 conditions. Metabolic changes associated with SLC25A38 loss suggest impaired mitochondrial pyridoxal 5'-phosphate-dependent enzymatic reactions, including serine to glycine conversion catalyzed by serine hydroxymethyltransferase-2 as well as ornithine aminotransferase. The proliferation defect of SLC25A38-null K562 cells in physiological and low vitamin B6 media can be explained by the loss of serine hydroxymethyltransferase-2-dependent production of one-carbon units and downstream de novo nucleotide synthesis. Our work points to a role for SLC25A38 in mitochondrial pyridoxal 5'-phosphate accumulation and provides insights into the pathology of congenital sideroblastic anemia.
许多必需蛋白质需要磷酸吡哆醛(维生素B6的活性形式)作为其活性的辅因子。这些蛋白质包括对氨基酸代谢、一碳代谢、多胺合成、红细胞生成和神经递质代谢至关重要的酶。所有哺乳动物中三分之一的磷酸吡哆醛依赖性酶定位于线粒体;然而,参与调节哺乳动物线粒体中磷酸吡哆醛水平的分子机制仍然未知。在这项研究中,我们在红白血病细胞中进行了全基因组CRISPR干扰筛选,并结合细胞器代谢组学,以确定线粒体内膜蛋白SLC25A38是线粒体磷酸吡哆醛的调节因子。SLC25A38的缺失导致线粒体而非细胞内磷酸吡哆醛的耗竭,并在生理条件和低维生素B6条件下均损害细胞增殖。与SLC25A38缺失相关的代谢变化表明,线粒体中依赖磷酸吡哆醛的酶促反应受损,包括丝氨酸羟甲基转移酶-2催化的丝氨酸向甘氨酸的转化以及鸟氨酸转氨酶。在生理条件和低维生素B6培养基中,SLC25A38基因敲除的K562细胞的增殖缺陷可以通过丝氨酸羟甲基转移酶-2依赖的一碳单位生成减少和下游从头核苷酸合成受损来解释。我们的工作表明SLC25A38在调节线粒体磷酸吡哆醛积累中发挥作用,并为先天性铁粒幼细胞贫血的病理机制提供了见解。