Liao Shi-Xia, Zhang Lan-Ying, Shi Ling-Mei, Hu Huai-Yu, Gu Yan-Hui, Wang Ting-Hua, Ouyang Yao, Sun Peng-Peng
Department of Respiratory and Critical Care Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi, 563003, China.
ShenQi Ethnic Medicine College of Guizhou Medical University, Zunyi, 550000, China.
Sci Rep. 2025 Feb 1;15(1):4005. doi: 10.1038/s41598-025-87437-2.
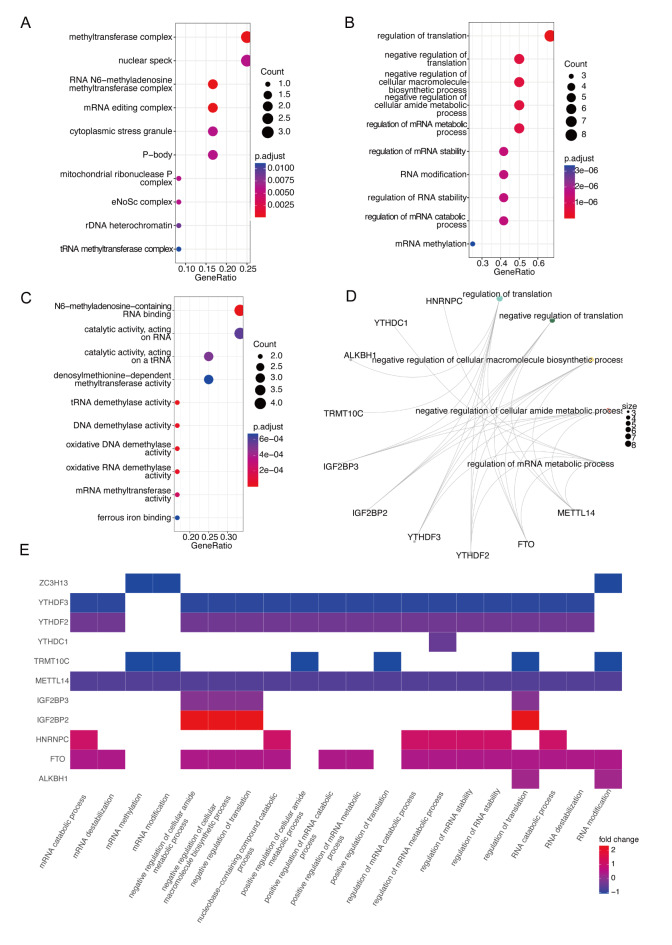
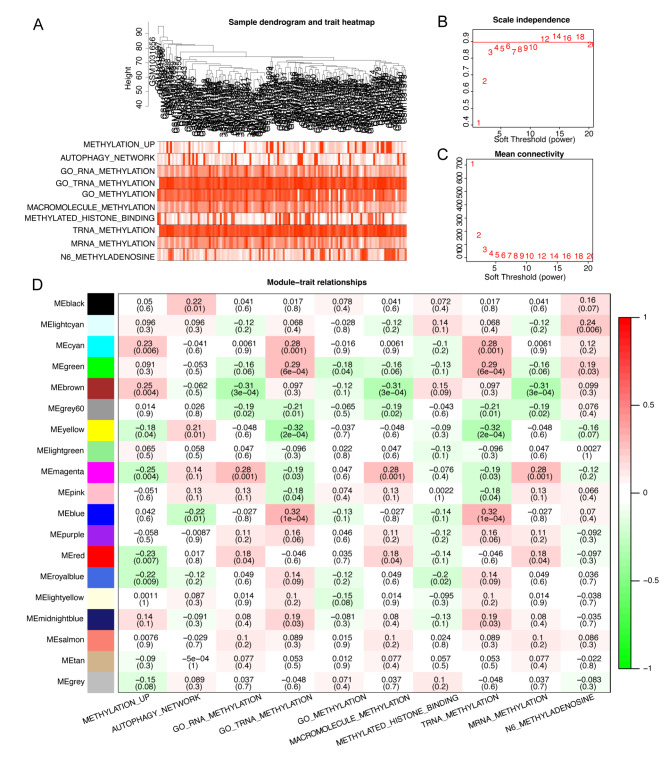
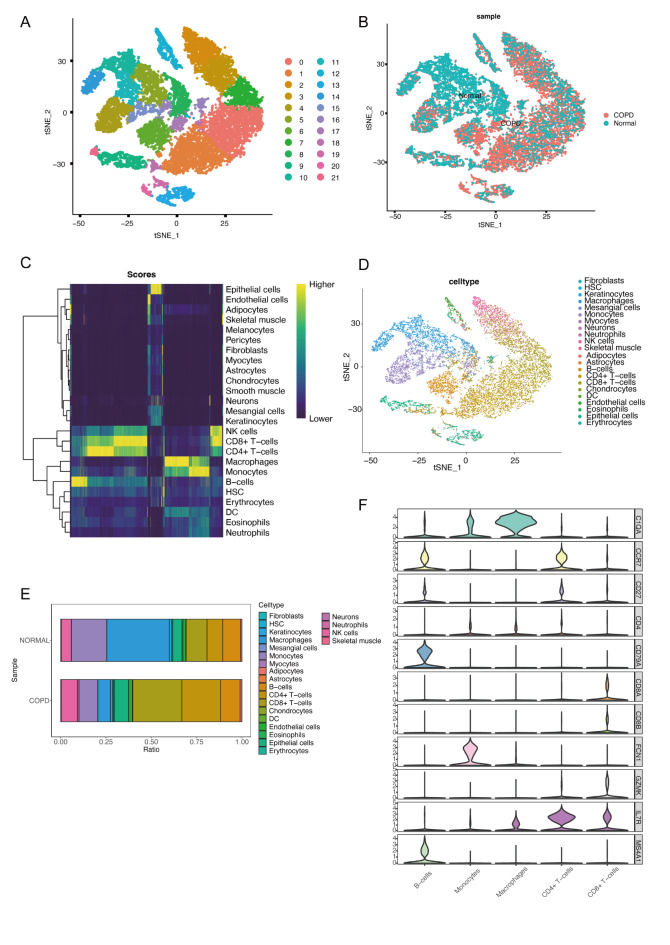
Chronic Obstructive Pulmonary Disease (COPD) is a heterogeneous lung disease influenced by epigenetic modifications, particularly RNA methylation. Emerging evidence also suggests that autophagy plays a crucial role in immune cell infiltration and is implicated in COPD progression. This study aimed to investigate key RNA methylation regulators and explore the roles of RNA methylation and autophagy in COPD pathogenesis. We analyzed tissue-based bulk RNA sequencing and single-cell RNA sequencing (scRNA-seq) datasets from COPD and non-COPD patients, sourced from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) were identified between COPD and non-COPD samples, and protein-protein interaction networks were constructed. Univariate logistic regression identified shared genes between DEGs and RNA methylation gene sets. Functional enrichment analyses, including Gene Ontology (GO), gene set enrichment analysis (GSEA), and gene set variation analysis (GSVA), were performed. Weighted gene co-expression network analysis (WGCNA) and immune infiltration analysis were conducted. Integration with scRNA-seq data further elucidated changes in immune cell composition, and cell communication analysis assessed interactions between macrophages and other immune cells. AddModuleScore analysis quantified RNA methylation and autophagy effects. Finally, a COPD mouse model was used to validate the expression of critical RNA methylation genes (FTO and IGF2BP2) in lung macrophages via RT-qPCR and flow cytometry. As revealed, we identified 13 RNA methylation-related genes enriched in translation and methylation processes. GSEA and GSVA revealed significant enrichment of these genes in immune and autophagy pathways. WGCNA analysis pinpointed key hub genes linking RNA methylation and autophagy. Integrated scRNA-seq analysis demonstrated a marked reduction of macrophages in COPD, with FTO and IGF2BP2 emerging as critical RNA methylation regulators. Macrophages with elevated RNA methylation and autophagy scores had increased interactions with other immune cells. In COPD mouse models, decreased expression of FTO and IGF2BP2 in lung macrophages was validated. Taken together, this study highlights the significant roles of RNA methylation in relation to autophagy pathways in the context of COPD. We identified key RNA methylation-related hub genes, such as FTO and IGF2BP2, which were found to have decreased expression in COPD macrophages. These findings provide novel genetic insights into the epigenetic mechanisms of COPD and suggest potential avenues for developing diagnostic and therapeutic strategies.
慢性阻塞性肺疾病(COPD)是一种受表观遗传修饰影响的异质性肺部疾病,尤其是RNA甲基化。新出现的证据还表明,自噬在免疫细胞浸润中起关键作用,并与COPD的进展有关。本研究旨在调查关键的RNA甲基化调节因子,并探讨RNA甲基化和自噬在COPD发病机制中的作用。我们分析了来自慢性阻塞性肺疾病患者和非慢性阻塞性肺疾病患者的基于组织的批量RNA测序和单细胞RNA测序(scRNA-seq)数据集,这些数据集来自基因表达综合数据库(GEO)。确定了慢性阻塞性肺疾病和非慢性阻塞性肺疾病样本之间的差异表达基因(DEGs),并构建了蛋白质-蛋白质相互作用网络。单变量逻辑回归确定了差异表达基因和RNA甲基化基因集之间的共享基因。进行了功能富集分析,包括基因本体(GO)、基因集富集分析(GSEA)和基因集变异分析(GSVA)。进行了加权基因共表达网络分析(WGCNA)和免疫浸润分析。与scRNA-seq数据的整合进一步阐明了免疫细胞组成的变化,细胞通讯分析评估了巨噬细胞与其他免疫细胞之间的相互作用。AddModuleScore分析量化了RNA甲基化和自噬效应。最后,使用慢性阻塞性肺疾病小鼠模型通过RT-qPCR和流式细胞术验证肺巨噬细胞中关键RNA甲基化基因(FTO和IGF2BP2)的表达。结果显示,我们确定了13个与RNA甲基化相关的基因,这些基因在翻译和甲基化过程中富集。GSEA和GSVA显示这些基因在免疫和自噬途径中显著富集。WGCNA分析确定了连接RNA甲基化和自噬的关键枢纽基因。综合scRNA-seq分析表明,慢性阻塞性肺疾病中巨噬细胞明显减少,FTO和IGF2BP2成为关键的RNA甲基化调节因子。RNA甲基化和自噬评分升高的巨噬细胞与其他免疫细胞的相互作用增加。在慢性阻塞性肺疾病小鼠模型中,验证了肺巨噬细胞中FTO和IGF2BP2的表达降低。综上所述,本研究强调了在慢性阻塞性肺疾病背景下RNA甲基化与自噬途径相关的重要作用。我们确定了关键的RNA甲基化相关枢纽基因,如FTO和IGF2BP2,发现它们在慢性阻塞性肺疾病巨噬细胞中的表达降低。这些发现为慢性阻塞性肺疾病的表观遗传机制提供了新的遗传学见解,并为开发诊断和治疗策略提供了潜在途径。