Zhang Chao, Nielsen Rasmus
Globe Institute, University of Copenhagen, Øster Voldgade 5-7, Copenhagen, 1350, Denmark.
Department of Integrative Biology and Department of Statistics, University of California Berkeley, 110 Sproul Hall, Berkeley, 94704, CA, USA.
bioRxiv. 2025 Jan 24:2025.01.20.633983. doi: 10.1101/2025.01.20.633983.
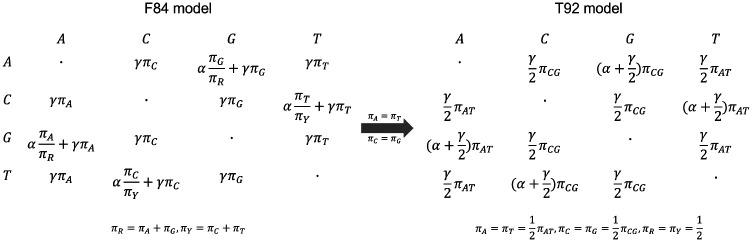
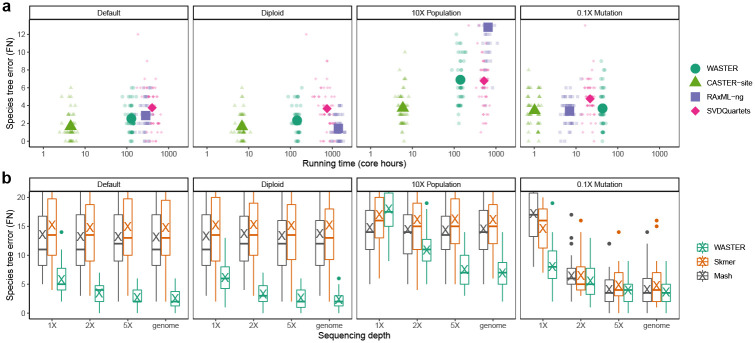
The advent of affordable whole-genome sequencing has spurred numerous large-scale projects aimed at inferring the tree of life, yet achieving a complete species-level phylogeny remains a distant goal due to significant costs and computational demands. Traditional species tree inference methods, though effective, are hampered by the need for high-coverage sequencing, high-quality genomic alignments, and extensive computational resources. To address these challenges, this study introduces WASTER, a novel tool for inferring species trees directly from short-read sequences. WASTER employs a k-mer based approach for identifying variable sites, circumventing the need for genome assembly and alignment. Using simulations, we demonstrate that WASTER achieves accuracy comparable to that of traditional alignment-based methods, even for low sequencing depth, and has substantially higher accuracy than other alignment-free methods. We validate WASTER's efficacy on real data, where it accurately reconstructs phylogenies of eukaryotic species with as low depth as 1.5X. WASTER provides a fast and efficient solution for phylogeny estimation in cases where genome assembly and/or alignment may bias analyses or is challenging, for example due to low sequencing depth. It also provides a method for generating guide trees for tree-based alignment algorithms. WASTER's ability to accurately estimate trees from low-coverage sequencing data without relying on assembly and alignment will lead to substantially reduced sequencing and computational costs in phylogenomic projects.
价格亲民的全基因组测序技术的出现推动了众多旨在推断生命树的大型项目,但由于成本高昂和计算需求巨大,要实现完整的物种水平系统发育仍然是一个遥不可及的目标。传统的物种树推断方法虽然有效,但因需要高覆盖率测序、高质量的基因组比对以及大量计算资源而受到限制。为应对这些挑战,本研究引入了WASTER,这是一种直接从短读序列推断物种树的新型工具。WASTER采用基于k-mer的方法来识别可变位点,无需进行基因组组装和比对。通过模拟,我们证明即使在低测序深度的情况下,WASTER的准确性也与传统的基于比对的方法相当,并且比其他无比对方法的准确性高得多。我们在真实数据上验证了WASTER的有效性,它能够准确重建深度低至1.5X的真核生物物种的系统发育。在基因组组装和/或比对可能会使分析产生偏差或具有挑战性的情况下,例如由于测序深度低,WASTER为系统发育估计提供了一种快速有效的解决方案。它还提供了一种为基于树的比对算法生成引导树的方法。WASTER能够在不依赖组装和比对的情况下从低覆盖率测序数据中准确估计树,这将大幅降低系统发育基因组学项目中的测序和计算成本。