Deng Yun, Song Yun S, Nielsen Rasmus
Center for Computational Biology, University of California, Berkeley, USA.
Department of Statistics, University of California, Berkeley, USA.
bioRxiv. 2025 Feb 15:2025.02.14.638385. doi: 10.1101/2025.02.14.638385.
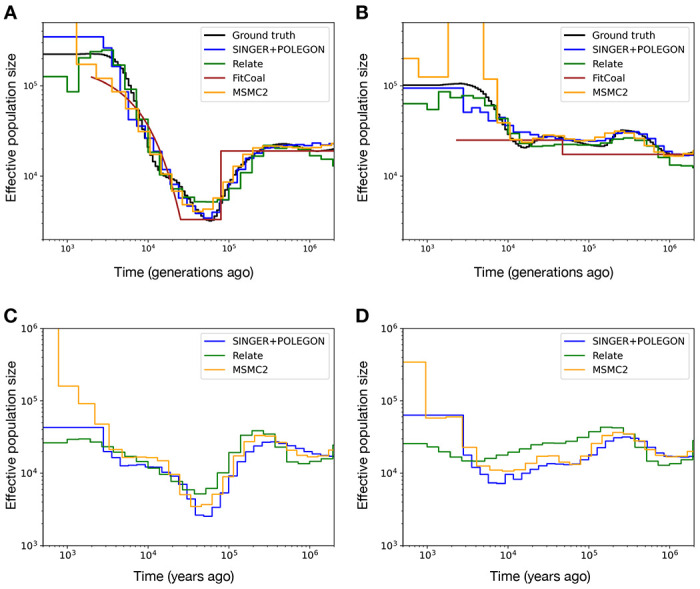
Inference of Ancestral Recombination Graphs (ARGs) is of central interest in the analysis of genomic variation. ARGs can be specified in terms of topologies and coalescence times. The coalescence times are usually estimated using an informative prior derived from coalescent theory, but this may generate biased estimates and can also complicate downstream inferences based on ARGs. Here we introduce, POLEGON, a novel approach for estimating branch lengths for ARGs which uses an uninformative prior. Using extensive simulations, we show that this method provides improved estimates of coalescence times and lead to more accurate inferences of effective population sizes under a wide range of demographic assumptions. It also improves other downstream inferences including estimates of mutation rates. We apply the method to data from the 1000 Genomes Project to investigate population size histories and differential mutation signatures across populations. We also estimate coalescence times in the HLA region, and show that they exceed 30 million years in multiple segments.
推断祖先重组图(ARG)是基因组变异分析的核心关注点。ARG可以根据拓扑结构和合并时间来指定。合并时间通常使用从合并理论推导出来的信息性先验进行估计,但这可能会产生有偏差的估计,并且还会使基于ARG的下游推断变得复杂。在这里,我们介绍了一种名为POLEGON的新颖方法,用于估计ARG的分支长度,该方法使用非信息性先验。通过广泛的模拟,我们表明该方法能够改进合并时间的估计,并在广泛的人口统计学假设下得出更准确的有效种群大小推断。它还改进了包括突变率估计在内的其他下游推断。我们将该方法应用于千人基因组计划的数据,以研究种群大小历史和不同种群间的差异突变特征。我们还估计了HLA区域的合并时间,并表明在多个片段中它们超过了3000万年。