Wan Xiaofeng, Zhan Jianmin, Ye Shuke, Chen Chuanrong, Li Runsheng, Shen Ming
National Health Commission (NHC) Key Lab of Reproduction Regulation (Shanghai Institute for Biomedical and Pharmaceutical Technologies), Shanghai, China.
Department of Oncology, Yijishan Hospital of Wannan Medical College, Wuhu, China.
Transl Cancer Res. 2025 Feb 28;14(2):930-948. doi: 10.21037/tcr-24-838. Epub 2025 Feb 24.
Breast cancer (BC) is a common tumor among women and is a heterogeneous disease with many subtypes. Each subtype shows different clinical presentations, disease trajectories and prognoses, and different responses to neoadjuvant therapy; thus, a new and universal prognostic biomarker for BC patients is urgently needed. Our goal is to identify a novel prognostic molecular biomarker that can accurately predict the outcome of all BC subtypes and guide their clinical management.
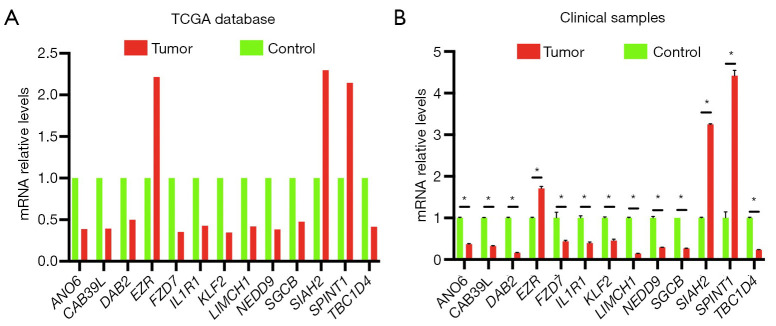
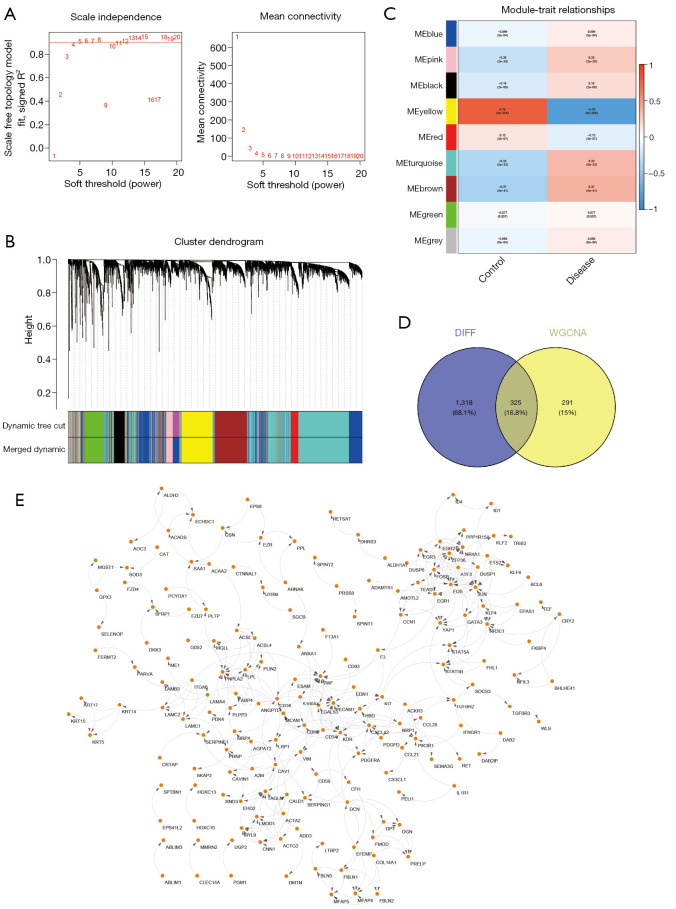
Utilizing data from The Cancer Genome Atlas (TCGA), we analyzed differential gene expression and patient clinical data. Weighted gene coexpression network analysis (WGCNA), Cox univariate regression and least absolute shrinkage and selection operator (LASSO) analysis were used to construct a prognostic model; the differential expression of the core genes in this model was validated via real-time quantitative polymerase chain reaction (RT-qPCR), and the reliability of the predictive model was validated in both an internal cohort and a BC patient dataset from the Gene Expression Omnibus (GEO) database. Further studies, such as gene set variation analysis (GSVA) and gene set enrichment analysis (GSEA), were performed to investigate the enrichment of signaling pathways. The CIBERSORT algorithm was used to estimate immune infiltration and tumor mutation burden (TMB), and drug sensitivity analysis was performed to evaluate the treatment response.
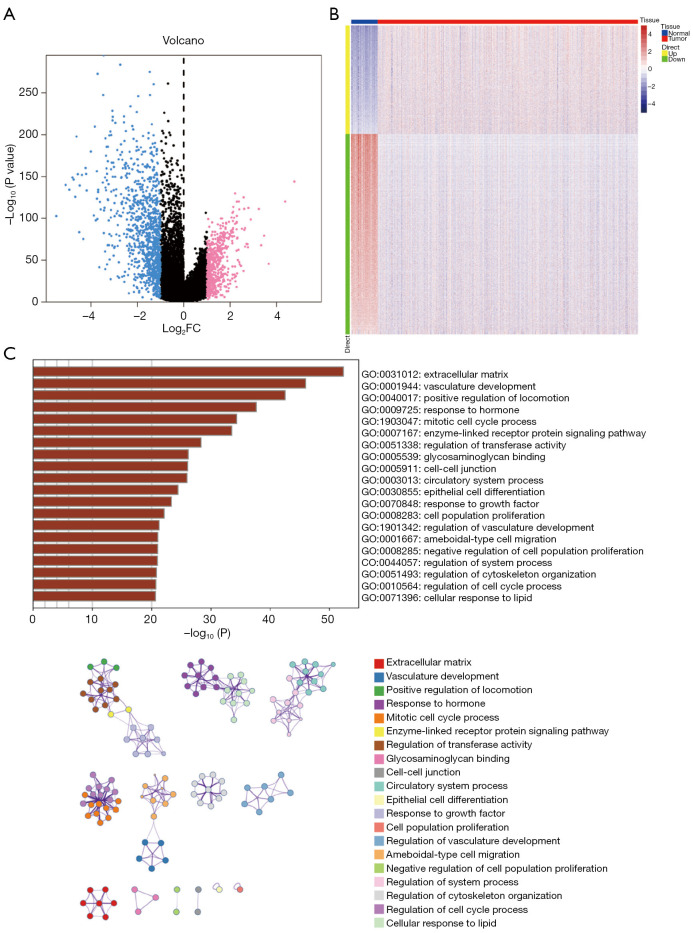
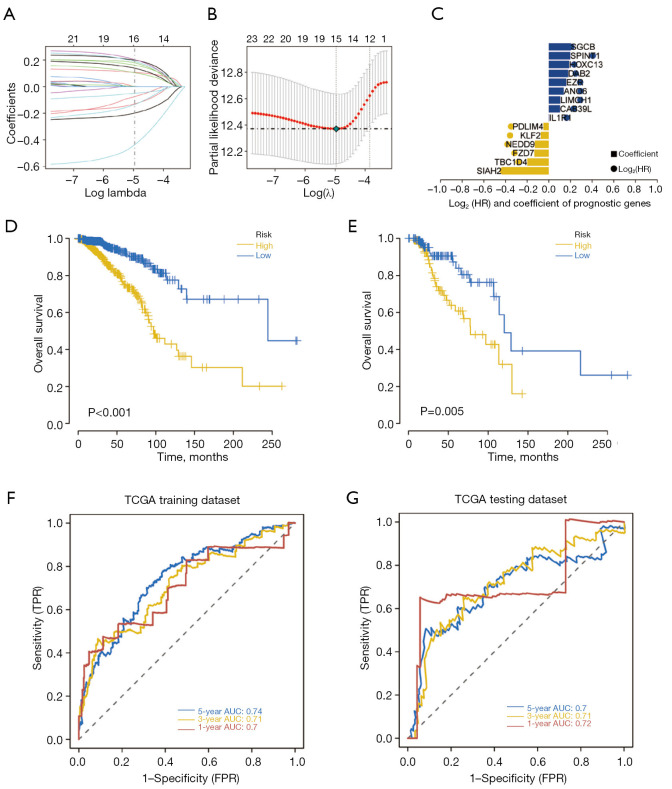
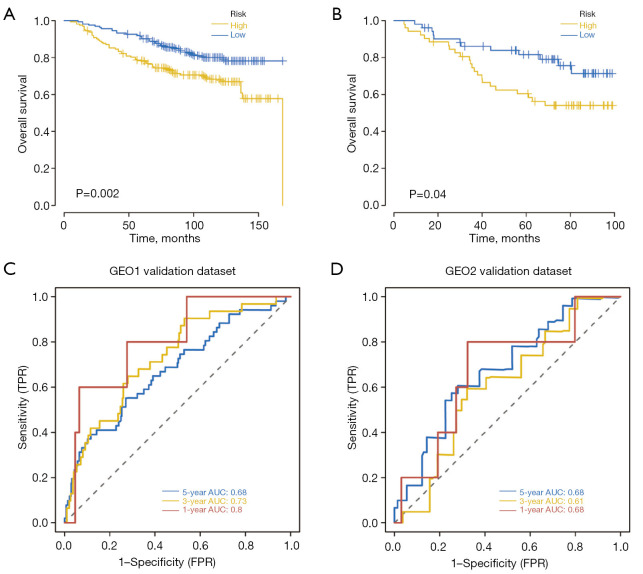
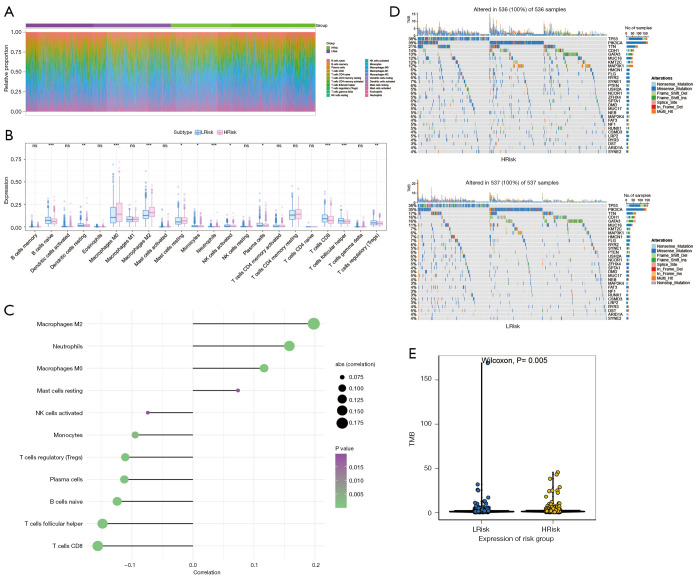
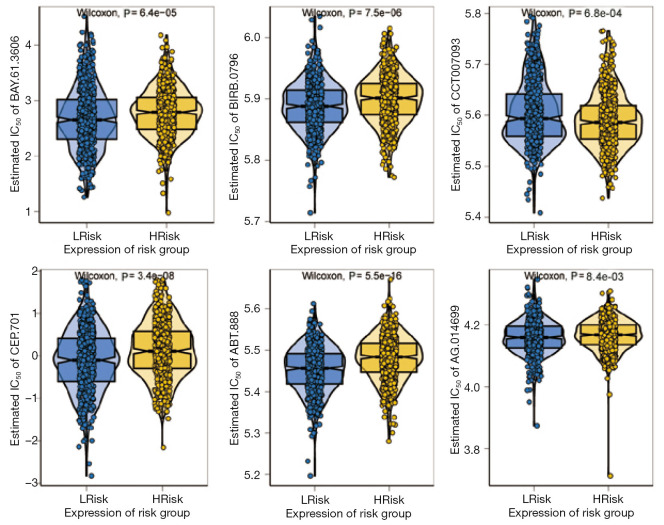
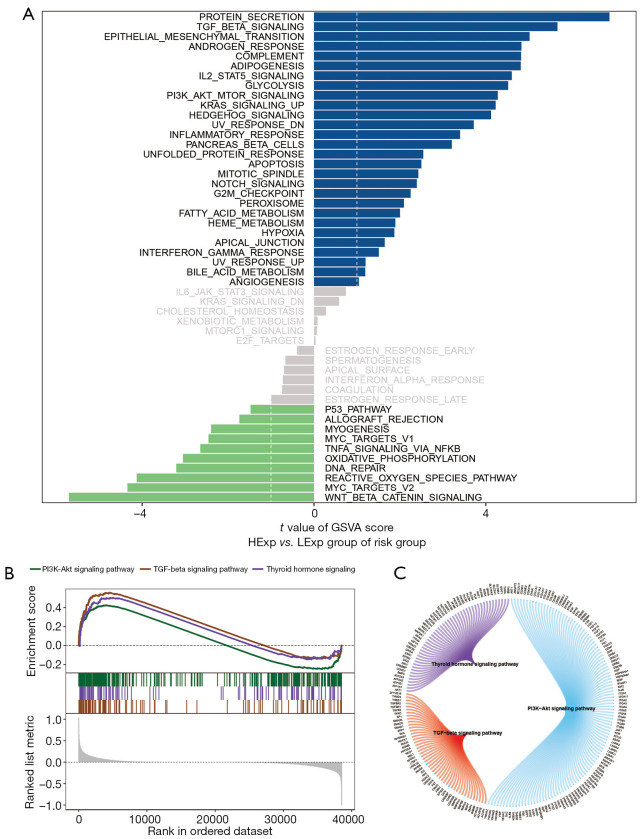
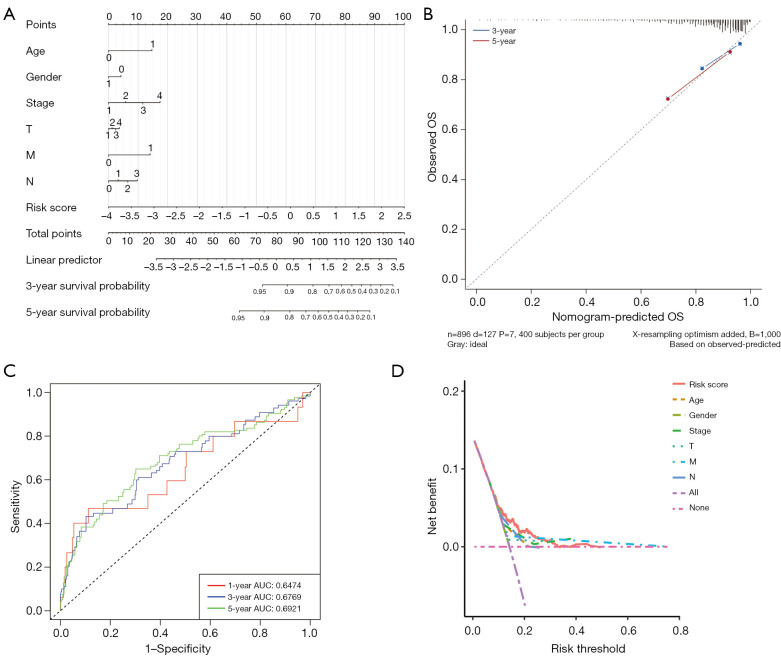
A total of 1,643 differentially expressed genes were identified. After WGCNA and Cox regression combined with LASSO analysis, 15 genes were identified by screening and used to establish a prognostic gene signature. Further analysis revealed that the epithelial-mesenchymal transition (EMT) pathway gene signature was enriched in these genes. Each patient was assigned a risk score, and according to the median risk score, patients were classified into a high-risk group or a low-risk group. The prognosis of the low-risk group was better than that of the high-risk group (P<0.01), and analyses of two independent GEO validation cohorts yielded similar results. Furthermore, a nomogram was constructed and found to perform well in predicting prognosis. GSVA revealed that the EMT pathway, transforming growth factor β (TGF-β) signaling pathway and PI3K-Akt signaling pathway genes were enriched in the high-risk group, and the Wnt-β-catenin signaling pathway, DNA repair pathway and P53 pathway gene sets were enriched in the low-risk group. GSEA revealed genes related to TGF-β signaling and the PI3K-Akt signaling pathways were enriched in the high-risk group. CIBERSORT demonstrated that the low-risk group had greater infiltration of antitumor immune cells. The TMB and drug sensitivity results suggested that immunotherapy and chemotherapy are likely to be more effective in the low-risk group.
We established a new EMT pathway-related prognostic gene signature that can be used to effectively predict BC prognosis and treatment response.
乳腺癌(BC)是女性常见的肿瘤,是一种具有多种亚型的异质性疾病。每种亚型表现出不同的临床表现、疾病轨迹和预后,以及对新辅助治疗的不同反应;因此,迫切需要一种新的、通用的BC患者预后生物标志物。我们的目标是鉴定一种新型预后分子生物标志物,它可以准确预测所有BC亚型的预后并指导其临床管理。
利用来自癌症基因组图谱(TCGA)的数据,我们分析了差异基因表达和患者临床数据。使用加权基因共表达网络分析(WGCNA)、Cox单变量回归和最小绝对收缩和选择算子(LASSO)分析构建预后模型;通过实时定量聚合酶链反应(RT-qPCR)验证该模型中核心基因的差异表达,并在内部队列和来自基因表达综合数据库(GEO)的BC患者数据集中验证预测模型的可靠性。进行了进一步的研究,如基因集变异分析(GSVA)和基因集富集分析(GSEA),以研究信号通路的富集情况。使用CIBERSORT算法估计免疫浸润和肿瘤突变负担(TMB),并进行药物敏感性分析以评估治疗反应。
共鉴定出1643个差异表达基因。经过WGCNA和Cox回归结合LASSO分析,筛选出15个基因并用于建立预后基因特征。进一步分析显示,上皮-间质转化(EMT)通路基因特征在这些基因中富集。为每位患者分配一个风险评分,并根据中位风险评分将患者分为高风险组或低风险组。低风险组的预后优于高风险组(P<0.01),对两个独立的GEO验证队列的分析也得出了类似的结果。此外,构建了一个列线图,发现其在预测预后方面表现良好。GSVA显示,EMT通路、转化生长因子β(TGF-β)信号通路和PI3K-Akt信号通路基因在高风险组中富集,而Wnt-β-连环蛋白信号通路、DNA修复通路和P53通路基因集在低风险组中富集。GSEA显示,与TGF-β信号通路和PI3K-Akt信号通路相关的基因在高风险组中富集。CIBERSORT表明,低风险组具有更强的抗肿瘤免疫细胞浸润。TMB和药物敏感性结果表明,免疫治疗和化疗在低风险组中可能更有效。
我们建立了一种新的与EMT通路相关的预后基因特征,可用于有效预测BC的预后和治疗反应。