Messingschlager Marey, Mackowiak Sebastian D, Voelker Maria Theresa, Bieg Matthias, Loske Jennifer, Chua Robert Lorenz, Liebig Johannes, Lukassen Sören, Thürmann Loreen, Seegebarth Anke, Twardziok Sven, Doncevic Daria, Herrmann Carl, Lorenz Stephan, Klages Sven, Steinbeis Fridolin, Witzenrath Martin, Kurth Florian, Conrad Christian, Sander Leif E, Ishaque Naveed, Eils Roland, Lehmann Irina, Laudi Sven, Trump Saskia
Berlin Institute of Health at Charité-Universitätsmedizin Berlin, Center of Digital Health, Molecular Epidemiology Unit, Berlin, Germany.
Freie Universität Berlin, Institute of Biology, Berlin, Germany.
EMBO Mol Med. 2025 May;17(5):923-937. doi: 10.1038/s44321-025-00215-5. Epub 2025 Mar 21.
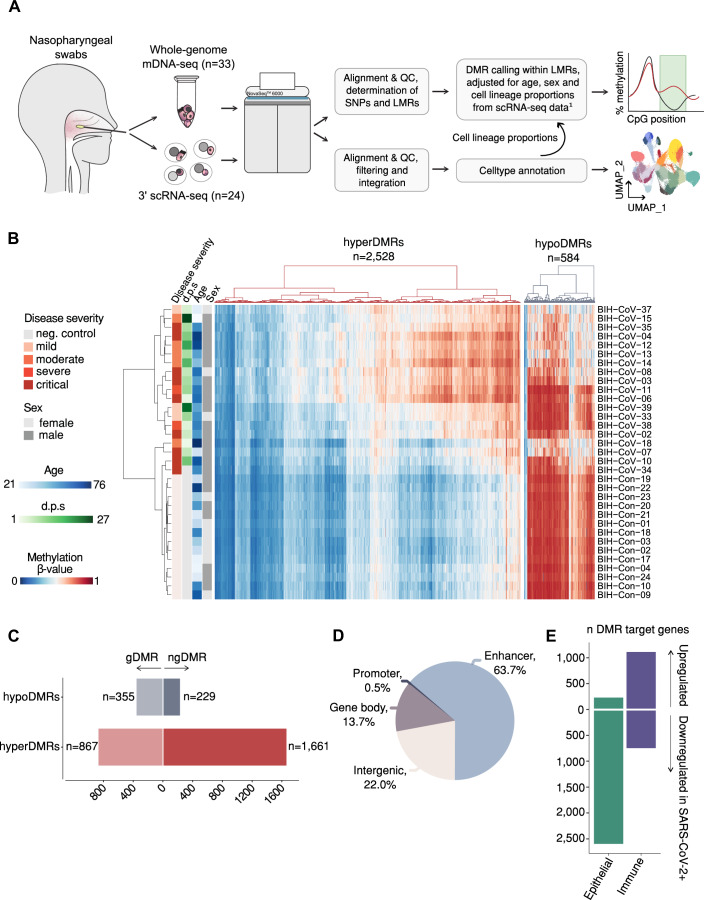
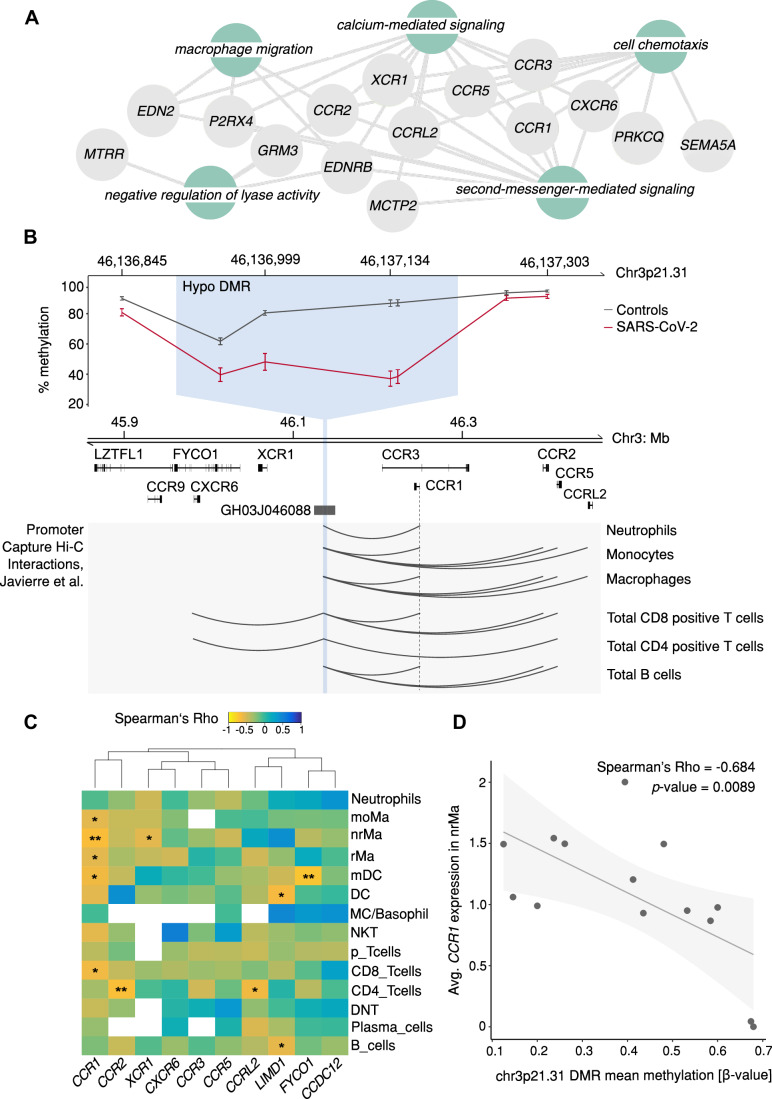
Molecular changes underlying the persistent health effects after SARS-CoV-2 infection remain poorly understood. To discern the gene regulatory landscape in the upper respiratory tract of COVID-19 patients, we performed enzymatic DNA methylome and single-cell RNA sequencing in nasal cells of COVID-19 patients (n = 19, scRNA-seq n = 14) and controls (n = 14, scRNA-seq n = 10). In addition, we resampled a subset of these patients for transcriptome analyses at 3 (n = 7) and 12 months (n = 5) post infection and followed the expression of differentially regulated genes over time. Genome-wide DNA methylation analysis revealed 3112 differentially methylated regions between COVID-19 patients and controls. Hypomethylated regions affected immune regulatory genes, while hypermethylated regions were associated with genes governing ciliary function. These genes were not only downregulated in the acute phase of the disease but sustained repressed up to 12 months post infection in ciliated cells. Validation in an independent cohort collected 6 months post infection (n = 15) indicated symptom-dependent transcriptional repression of ciliary genes. We therefore propose that hypermethylation observed in the acute phase may exert a long-term effect on gene expression, possibly contributing to post-acute COVID-19 sequelae.
新冠病毒感染后持续健康影响背后的分子变化仍知之甚少。为了识别新冠患者上呼吸道中的基因调控格局,我们对新冠患者(n = 19,单细胞RNA测序n = 14)和对照组(n = 14,单细胞RNA测序n = 10)的鼻腔细胞进行了酶促DNA甲基化组和单细胞RNA测序。此外,我们对这些患者的一个子集在感染后3个月(n = 7)和12个月(n = 5)进行了重采样以进行转录组分析,并随时间追踪差异调节基因的表达。全基因组DNA甲基化分析揭示了新冠患者和对照组之间有3112个差异甲基化区域。低甲基化区域影响免疫调节基因,而高甲基化区域与控制纤毛功能的基因相关。这些基因不仅在疾病急性期下调,而且在纤毛细胞中直至感染后12个月仍持续受到抑制。在感染后6个月收集的一个独立队列(n = 15)中的验证表明,纤毛基因的转录抑制与症状有关。因此,我们提出在急性期观察到的高甲基化可能对基因表达产生长期影响,这可能导致新冠后后遗症。