Kaplun Ludmila, Krautz-Peterson Greice, Neerman Nir, Schindler Yocheved, Dehan Elinor, Huettner Claudia S, Baumgartner Brett K, Stanley Christine, Kaplun Alexander
Variantyx Inc., Framingham, MA 01701, USA.
Int J Mol Sci. 2025 Mar 18;26(6):2725. doi: 10.3390/ijms26062725.
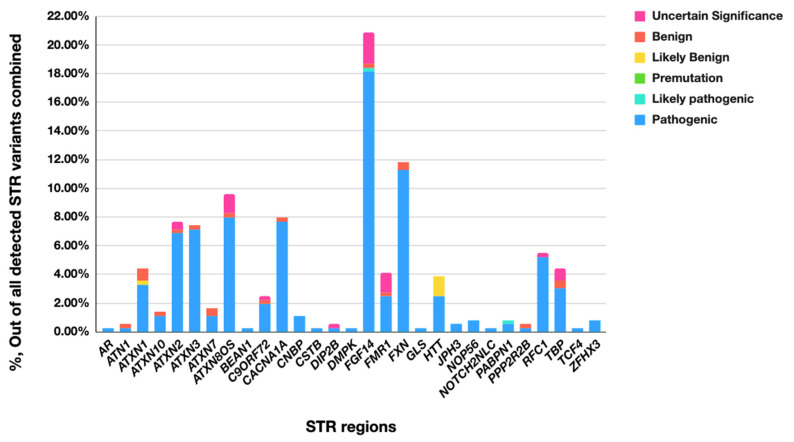
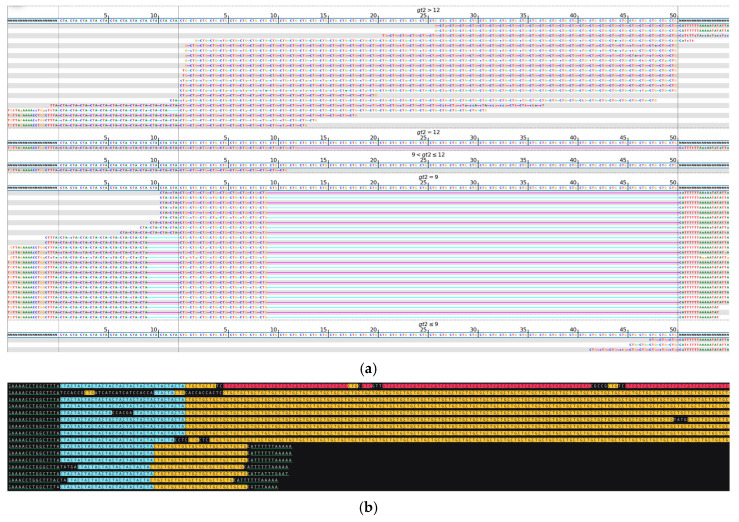
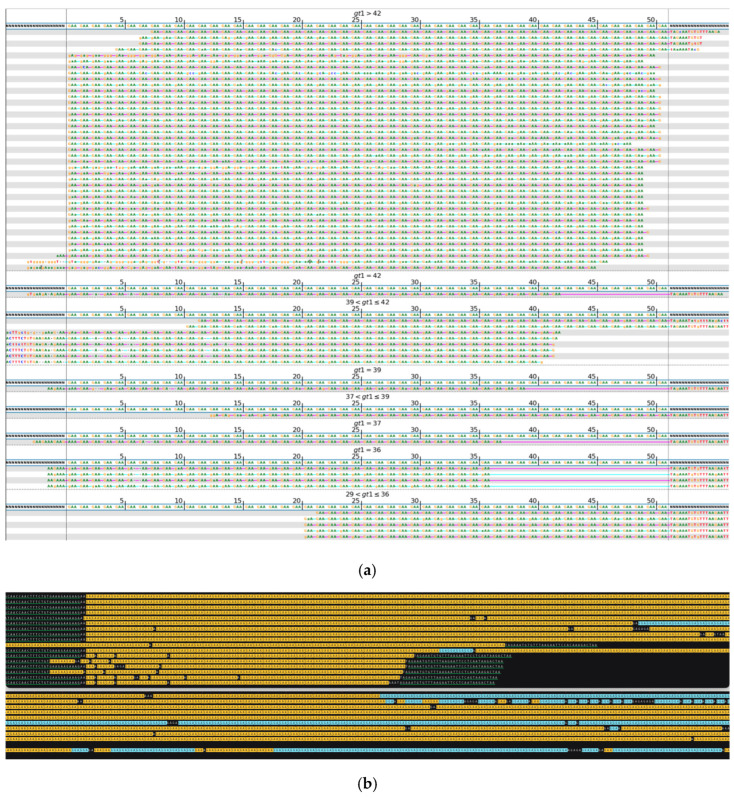
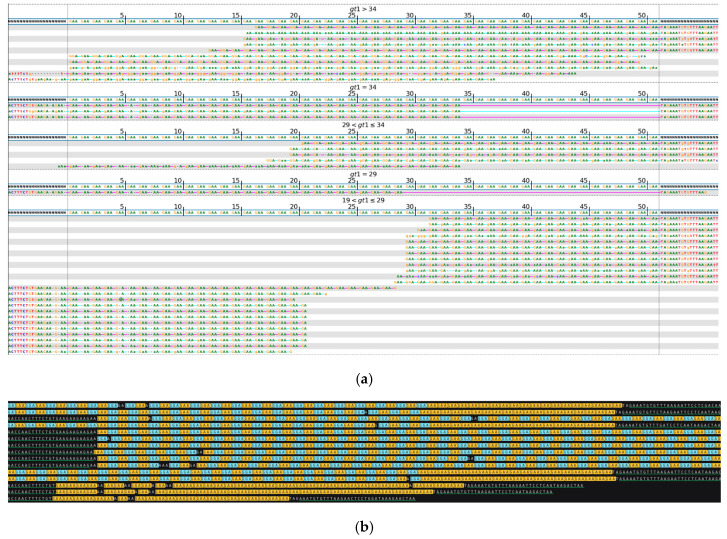
While whole-genome sequencing (WGS) using short-read technology has become a standard diagnostic test, this technology has limitations in analyzing certain genomic regions, particularly short tandem repeats (STRs). These repetitive sequences are associated with over 50 diseases, primarily affecting neurological function, including Huntington disease, frontotemporal dementia, and Friedreich's ataxia. We analyzed 2689 cases with movement disorders and dementia-related phenotypes processed at Variantyx in 2023-2024 using a two-tiered approach, with an initial short-read WGS followed by ONT long-read sequencing (when necessary) for variant characterization. Of the 2038 cases (75.8%) with clinically relevant genetic variants, 327 (16.0%) required additional long-read analysis. STR variants were reported in 338 cases (16.6% of positive cases), with approximately half requiring long-read sequencing for definitive classification. The combined approach enabled the precise determination of repeat length, composition, somatic mosaicism, and methylation status. Notable advantages included the detection of complex repeat structures in several genes such as , , and , where long-read sequencing allowed to determine somatic repeat unit variations and accurate allele phasing. Further studies are needed to establish technology-specific guidelines for the standardized interpretation of long-read sequencing data for the clinical diagnostics of repeat expansion disorders.
虽然使用短读长技术的全基因组测序(WGS)已成为标准诊断测试,但该技术在分析某些基因组区域(特别是短串联重复序列(STR))时存在局限性。这些重复序列与50多种疾病相关,主要影响神经功能,包括亨廷顿病、额颞叶痴呆和弗里德赖希共济失调。我们使用两层方法分析了2023年至2024年在Variantyx处理的2689例运动障碍和痴呆相关表型的病例,首先进行初始短读长WGS,必要时进行ONT长读长测序以进行变异特征分析。在2038例(75.8%)具有临床相关遗传变异的病例中,327例(16.0%)需要额外的长读长分析。338例(阳性病例的16.6%)报告了STR变异,其中约一半需要长读长测序进行明确分类。这种联合方法能够精确确定重复长度、组成、体细胞嵌合性和甲基化状态。显著优势包括在几个基因(如 、 和 )中检测到复杂的重复结构,长读长测序能够确定体细胞重复单元变异和准确的等位基因分型。需要进一步研究以建立技术特定的指南,用于对重复扩增疾病的临床诊断进行长读长测序数据的标准化解释。