Wang Min, Yuan Tingting, Chen Jiali, Yang Jing, Pu Ji, Lin Wenchao, Dong Kui, Zhang Luqing, Yuan Jiale, Zheng Han, Sun Yamin, Xu Jianguo
National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases, TEDA Institute of Biological Sciences and Biotechnology, Nankai University, Tianjin, China.
School of Medicine, Research Institute of Public Health, Nankai University, Tianjin, China.
Front Microbiol. 2025 Mar 28;16:1553124. doi: 10.3389/fmicb.2025.1553124. eCollection 2025.
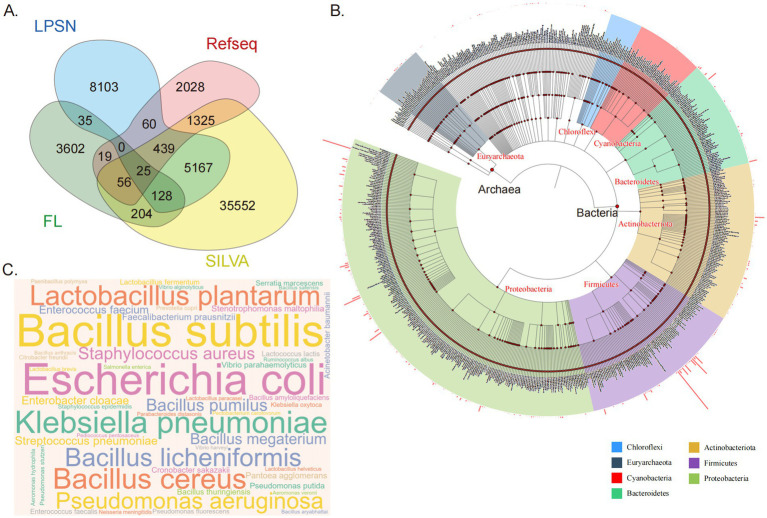
16S rRNA gene sequencing is pivotal for identifying bacterial species in microbiome studies, especially using the V3-V4 hypervariable regions. A fixed 98.5% similarity threshold is often applied for species-level identification, but this approach can cause misclassification due to varying thresholds among species. To address this, our study integrated data from SILVA, NCBI, and LPSN databases, extracting V3-V4 region sequences and supplementing them with 16S rRNA sequences from 1,082 human gut samples. This resulted in a non-redundant amplicon sequence variants (ASVs) database specific to the V3-V4 regions (positions 341-806). Utilizing this database, we identified flexible classification thresholds for 674 families, 3,661 genera, and 15,735 species, finding clear thresholds for 87.09% of families and 98.38% of genera. For the 896 most common human gut species, we established precise taxonomic thresholds. To leverage these findings, we developed the asvtax pipeline, which applies flexible thresholds for more accurate taxonomic classification, notably improving the identification of new ASVs. The asvtax pipeline not only enhances the precision of species-level classification but also provides a robust framework for analyzing complex microbial communities, facilitating more reliable ecological and functional interpretations in microbiome research.
16S rRNA基因测序对于微生物组研究中细菌物种的鉴定至关重要,尤其是使用V3 - V4高变区时。物种水平鉴定通常采用固定的98.5%相似度阈值,但由于不同物种间阈值存在差异,这种方法可能导致错误分类。为解决这一问题,我们的研究整合了来自SILVA、NCBI和LPSN数据库的数据,提取V3 - V4区域序列,并补充了来自1082份人类肠道样本的16S rRNA序列。这产生了一个特定于V3 - V4区域(位置341 - 806)的非冗余扩增子序列变体(ASV)数据库。利用该数据库,我们确定了674个科、3661个属和15735个物种的灵活分类阈值,发现87.09%的科和98.38%的属有明确阈值。对于896种最常见的人类肠道物种,我们建立了精确的分类阈值。为利用这些发现,我们开发了asvtax流程,该流程应用灵活阈值进行更准确的分类学分类,显著提高了新ASV的鉴定能力。asvtax流程不仅提高了物种水平分类的精度,还为分析复杂微生物群落提供了一个强大的框架,有助于在微生物组研究中进行更可靠的生态和功能解读。