Cellex Laboratory, CibeRes (Centro de Investigación Biomédica en Red de Enfermedades Respiratorias, 06/06/0028), Institut d'Investigacions Biomèdiques August Pi I Sunyer (IDIBAPS), Barcelona, Spain.
School of Medicine, University of Barcelona, Barcelona, Spain.
Sci Rep. 2023 Mar 9;13(1):3974. doi: 10.1038/s41598-023-30764-z.
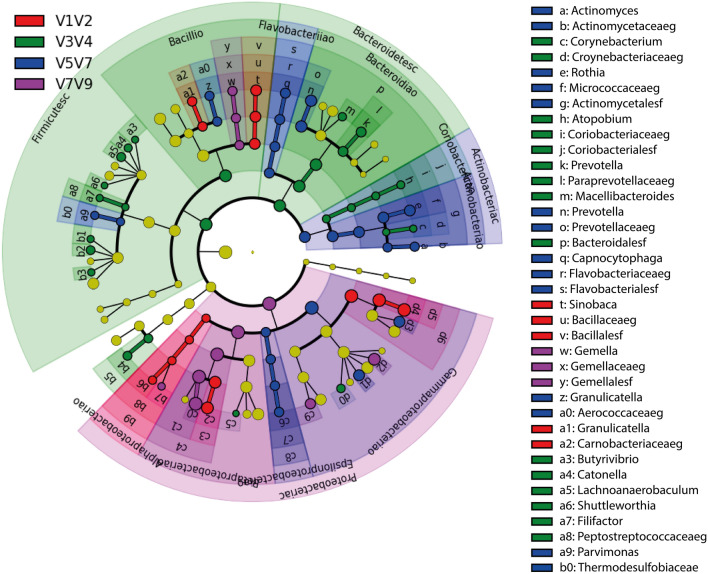
16S rRNA gene profiling, which contains nine hypervariable regions (V1-V9), is the gold standard for identifying taxonomic units by high-throughput sequencing. Microbiome studies combine two or more region sequences (usually V3-V4) to increase the resolving power for identifying bacterial taxa. We compare the resolving powers of V1-V2, V3-V4, V5-V7, and V7-V9 to improve microbiome analyses in sputum samples from patients with chronic respiratory diseases. DNA were isolated from 33 human sputum samples, and libraries were created using a QIASeq screening panel intended for Illumina platforms (16S/ITS; Qiagen Hilden, Germany). The analysis included a mock community as a microbial standard control (ZymoBIOMICS). We used the Deblur algorithm to identify bacterial amplicon sequence variants (ASVs) at the genus level. Alpha diversity was significantly higher for V1-V2, V3-V4, and V5-V7 compared with V7-V9, and significant compositional dissimilarities in the V1-V2 and V7-V9 analyses versus the V3-V4 and V5-V7 analyses. A cladogram confirmed these compositional differences, with the latter two being very similar in composition. The combined hypervariable regions showed significant differences when discriminating between the relative abundances of bacterial genera. The area under the curve revealed that V1-V2 had the highest resolving power for accurately identifying respiratory bacterial taxa from sputum samples. Our study confirms that 16S rRNA hypervariable regions provide significant differences for taxonomic identification in sputum. Comparing the taxa of microbial community standard control with the taxa samples, V1-V2 combination exhibits the most sensitivity and specificity. Thus, while third generation full-length 16S rRNA sequencing platforms become more available, the V1-V2 hypervariable regions can be used for taxonomic identification in sputum.
16S rRNA 基因谱分析,其中包含九个高变区(V1-V9),是通过高通量测序识别分类单元的金标准。微生物组研究结合两个或更多区域序列(通常为 V3-V4),以提高识别细菌分类群的分辨率。我们比较了 V1-V2、V3-V4、V5-V7 和 V7-V9 的分辨率,以改善慢性呼吸道疾病患者痰液样本的微生物组分析。从 33 个人类痰液样本中分离 DNA,并使用旨在用于 Illumina 平台的 QIASeq 筛选面板(16S/ITS;Qiagen Hilden,德国)创建文库。分析包括作为微生物标准对照的模拟群落(ZymoBIOMICS)。我们使用 Deblur 算法在属水平上识别细菌扩增子序列变体(ASV)。与 V7-V9 相比,V1-V2、V3-V4 和 V5-V7 的 alpha 多样性显著更高,并且 V1-V2 和 V7-V9 分析与 V3-V4 和 V5-V7 分析之间的组成差异显著。系统发育树证实了这些组成差异,后两者在组成上非常相似。组合高变区在区分细菌属的相对丰度方面显示出显著差异。曲线下面积表明,V1-V2 具有最高的分辨率,可从痰液样本中准确识别呼吸道细菌分类群。我们的研究证实,16S rRNA 高变区在痰液中提供了用于分类鉴定的显著差异。将微生物群落标准对照的分类群与样本分类群进行比较,V1-V2 组合表现出最高的灵敏度和特异性。因此,虽然第三代全长 16S rRNA 测序平台变得更加可用,但 V1-V2 高变区可用于痰液的分类鉴定。