Pivirotto Alyssa, Peles Noah, Hey Jody
Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Library & Information Technology Services, Bryn Mawr College, Bryn Mawr, PA 19010, USA.
G3 (Bethesda). 2025 Jun 4;15(6). doi: 10.1093/g3journal/jkaf088.
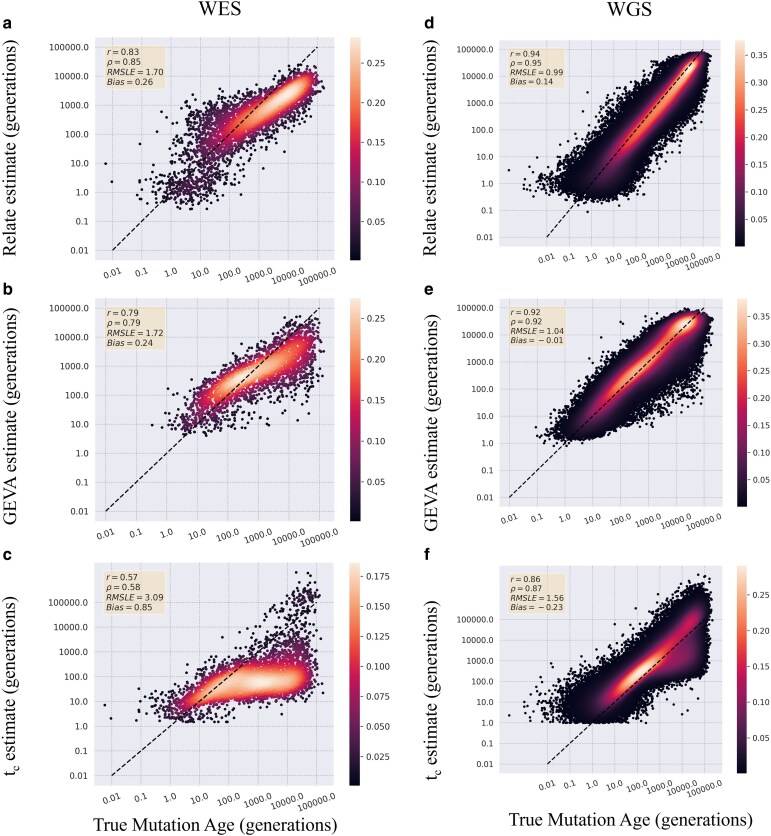
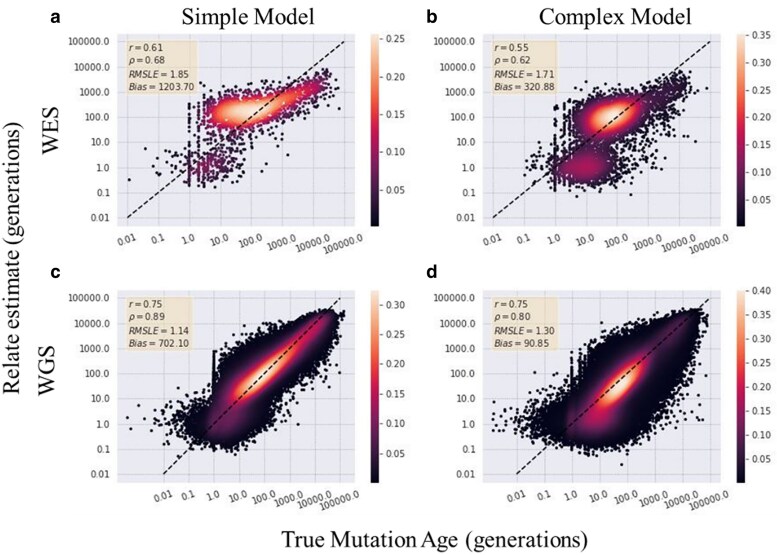
As personalized genomics becomes more affordable, larger numbers of rare variants are being discovered, leading to important initiatives in identifying the functional impacts in relation to disease phenotypes. One way to characterize these variants is to estimate the time the mutation entered the population. However, allele age estimators such as those implemented in the programs Relate, Genealogical Estimator of Variant Age, and Runtc were developed based on the assumption that datasets include the entire genome. We examined the performance of each of these estimators on simulated exome data under a neutral constant population size model, as well as under population expansion and background selection models. We found that each provides usable estimates of allele age from whole-exome datasets. Relate performs the best amongst all 3 estimators with Pearson coefficients of 0.83 and 0.73 (with respect to true simulated values for neutral constant and expansion population models, respectively) with a 12% and 20% decrease in correlation between whole-genome and whole-exome estimations. Of the 3 estimators, Relate is best able to parallelize to yield quick results with little resources; however, Relate is currently only able to scale to thousands of samples making it unable to match the hundreds of thousands of samples being currently released. While more work is needed to expand the capabilities of current methods of estimating allele age, these methods show a modest decrease in performance in the estimation of the age of mutations.
随着个性化基因组学的成本越来越低,人们发现了大量罕见变异,这引发了一些重要举措,旨在确定这些变异与疾病表型相关的功能影响。表征这些变异的一种方法是估计突变进入群体的时间。然而,诸如Relate、变异年龄谱系估计器和Runtc等程序中实现的等位基因年龄估计器是基于数据集包含整个基因组这一假设开发的。我们在中性恒定群体大小模型以及群体扩张和背景选择模型下,研究了这些估计器在模拟外显子组数据上的性能。我们发现,每个估计器都能从全外显子组数据集中提供可用的等位基因年龄估计。在所有3个估计器中,Relate的表现最佳,其皮尔逊系数分别为0.83和0.73(分别相对于中性恒定和扩张群体模型的真实模拟值),全基因组估计和全外显子组估计之间的相关性分别下降了12%和20%。在这3个估计器中,Relate最能够并行化以用很少的资源快速得出结果;然而,Relate目前只能扩展到数千个样本,这使其无法匹配当前发布的数十万个样本。虽然需要更多工作来扩展当前估计等位基因年龄方法的能力,但这些方法在估计突变年龄时性能仅略有下降。