Zhang Ren-Gang, Shang Hong-Yun, Milne Richard Ian, Almeida-Silva Fabricio, Chen Hengchi, Zhou Min-Jie, Shu Heng, Jia Kai-Hua, Van de Peer Yves, Ma Yong-Peng
State Key Laboratory of Plant Diversity and Specialty Crops/Yunnan Key Laboratory for Integrative Conservation of Plant Species with Extremely Small Populations, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China.
University of the Chinese Academy of Sciences, Beijing 101408, China.
Nucleic Acids Res. 2025 Apr 10;53(7). doi: 10.1093/nar/gkaf320.
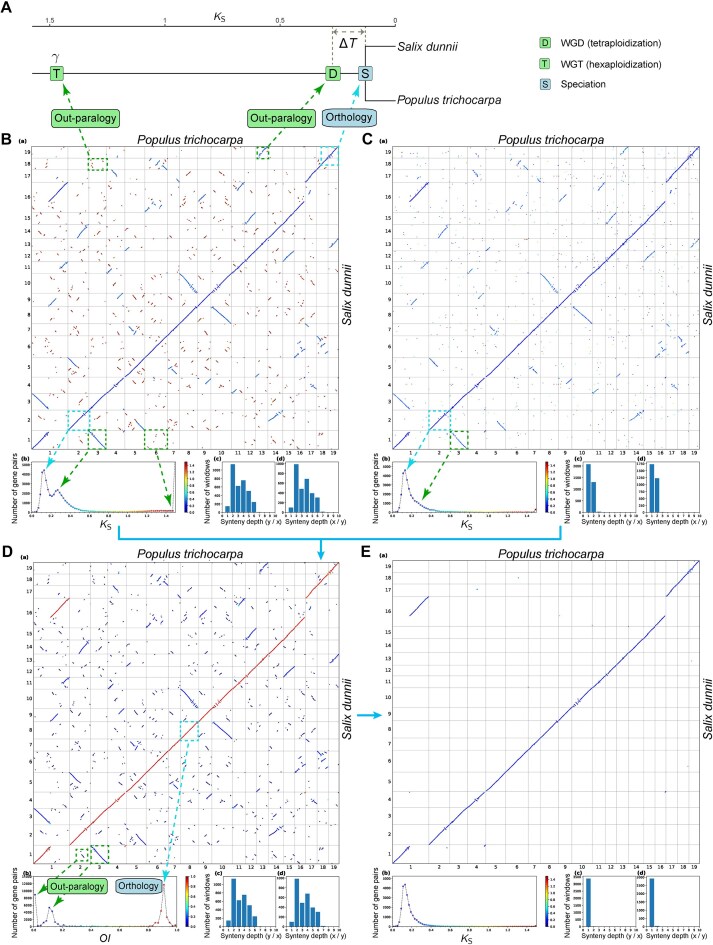
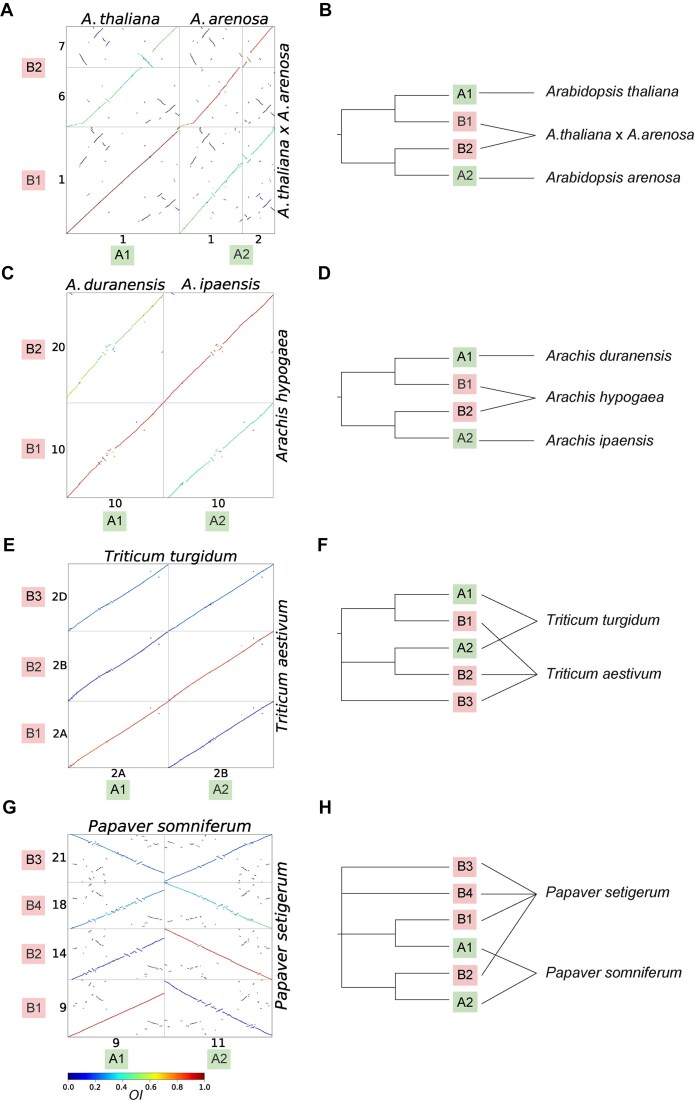
With the explosive growth of whole-genome datasets, accurate detection of orthologous synteny has become crucial for reconstructing evolutionary history. However, current methods for identifying orthologous synteny face great limitations, particularly in scaling with varied polyploidy histories and accurately removing out-paralogous synteny. In this study, we developed a scalable and robust approach, based on the Orthology Index (OI), to effectively identify orthologous synteny. Our evaluation across a large-scale empirical dataset with diverse polyploidization events demonstrated the high reliability and robustness of the OI method. Simulation-based benchmarks further validated the accuracy of our method, showing its superior performance against existing methods across a wide range of scenarios. Additionally, we explored its broad applications in reconstructing the evolutionary histories of plant genomes, including the inference of polyploidy, identification of reticulation, and phylogenomics. In conclusion, OI offers a robust, interpretable, and scalable approach for identifying orthologous synteny, facilitating more accurate and efficient analyses in plant evolutionary genomics.
随着全基因组数据集的爆炸式增长,准确检测直系同源同线性对于重建进化历史变得至关重要。然而,目前识别直系同源同线性的方法面临很大局限性,特别是在适应不同多倍体历史以及准确去除旁系同源同线性方面。在本研究中,我们基于直系同源指数(OI)开发了一种可扩展且稳健的方法,以有效识别直系同源同线性。我们对具有多样多倍化事件的大规模实证数据集进行的评估证明了OI方法的高可靠性和稳健性。基于模拟的基准测试进一步验证了我们方法的准确性,表明其在广泛场景下相对于现有方法具有卓越性能。此外,我们探索了其在重建植物基因组进化历史中的广泛应用,包括多倍体推断、网状化识别和系统发育基因组学。总之,OI为识别直系同源同线性提供了一种稳健、可解释且可扩展的方法,有助于在植物进化基因组学中进行更准确高效的分析。