Department of Plant and Microbial Biology, University of California, Berkeley, CA 94720, USA.
BMC Bioinformatics. 2011 Apr 18;12:102. doi: 10.1186/1471-2105-12-102.
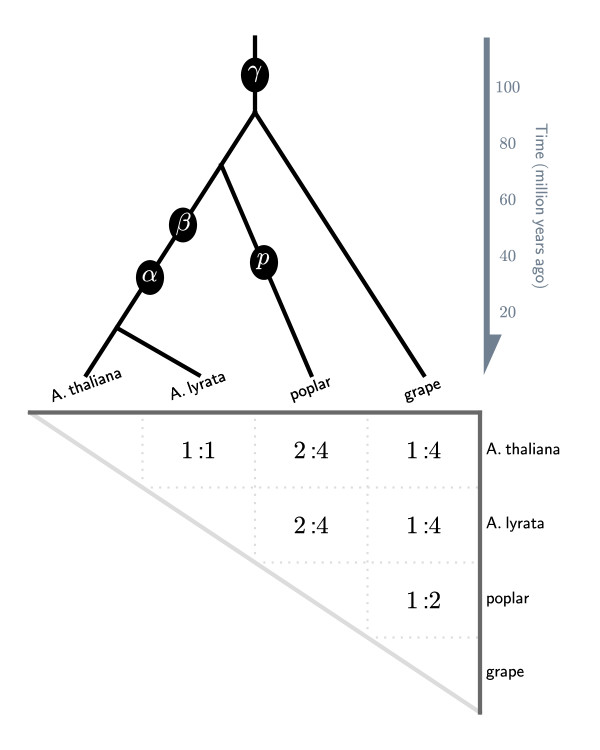
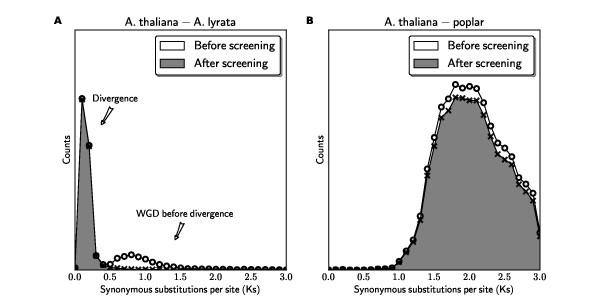
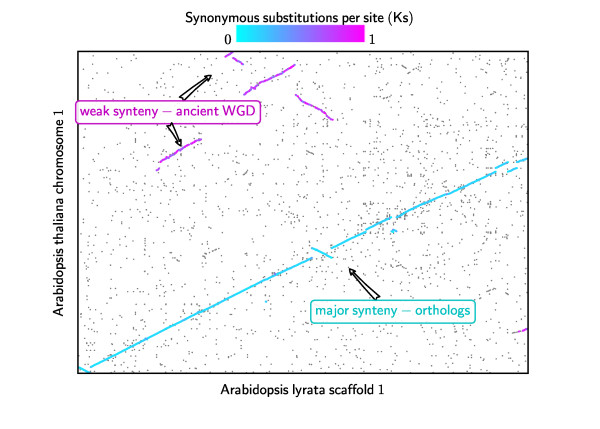
It is difficult to accurately interpret chromosomal correspondences such as true orthology and paralogy due to significant divergence of genomes from a common ancestor. Analyses are particularly problematic among lineages that have repeatedly experienced whole genome duplication (WGD) events. To compare multiple "subgenomes" derived from genome duplications, we need to relax the traditional requirements of "one-to-one" syntenic matchings of genomic regions in order to reflect "one-to-many" or more generally "many-to-many" matchings. However this relaxation may result in the identification of synteny blocks that are derived from ancient shared WGDs that are not of interest. For many downstream analyses, we need to eliminate weak, low scoring alignments from pairwise genome comparisons. Our goal is to objectively select subset of synteny blocks whose total scores are maximized while respecting the duplication history of the genomes in comparison. We call this "quota-based" screening of synteny blocks in order to appropriately fill a quota of syntenic relationships within one genome or between two genomes having WGD events.
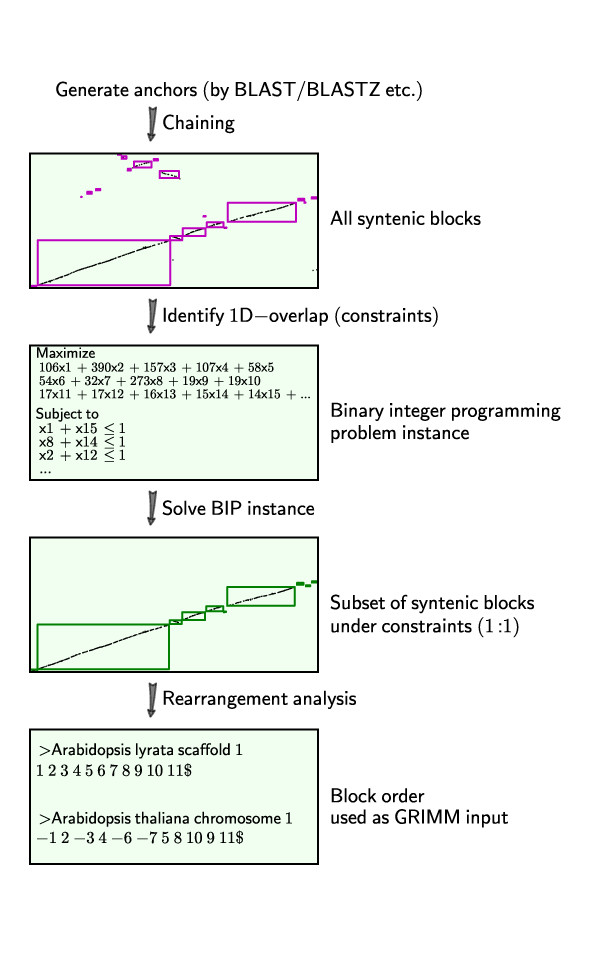
We have formulated the synteny block screening as an optimization problem known as "Binary Integer Programming" (BIP), which is solved using existing linear programming solvers. The computer program QUOTA-ALIGN performs this task by creating a clear objective function that maximizes the compatible set of synteny blocks under given constraints on overlaps and depths (corresponding to the duplication history in respective genomes). Such a procedure is useful for any pairwise synteny alignments, but is most useful in lineages affected by multiple WGDs, like plants or fish lineages. For example, there should be a 1:2 ploidy relationship between genome A and B if genome B had an independent WGD subsequent to the divergence of the two genomes. We show through simulations and real examples using plant genomes in the rosid superorder that the quota-based screening can eliminate ambiguous synteny blocks and focus on specific genomic evolutionary events, like the divergence of lineages (in cross-species comparisons) and the most recent WGD (in self comparisons).
The QUOTA-ALIGN algorithm screens a set of synteny blocks to retain only those compatible with a user specified ploidy relationship between two genomes. These blocks, in turn, may be used for additional downstream analyses such as identifying true orthologous regions in interspecific comparisons. There are two major contributions of QUOTA-ALIGN: 1) reducing the block screening task to a BIP problem, which is novel; 2) providing an efficient software pipeline starting from all-against-all BLAST to the screened synteny blocks with dot plot visualizations. Python codes and full documentations are publicly available http://github.com/tanghaibao/quota-alignment. QUOTA-ALIGN program is also integrated as a major component in SynMap http://genomevolution.com/CoGe/SynMap.pl, offering easier access to thousands of genomes for non-programmers.
由于与共同祖先的基因组存在显著差异,因此准确解释染色体对应关系(如真正的同源和旁系同源)具有一定难度。在经历了多次全基因组复制(WGD)事件的谱系中,分析尤其成问题。为了比较来自基因组复制的多个“亚基因组”,我们需要放宽对基因组区域“一一”同线性匹配的传统要求,以反映“一对多”或更一般地“多对多”匹配。但是,这种放宽可能会导致识别出源自非感兴趣的古老共享 WGD 的同线性块。对于许多下游分析,我们需要从两两基因组比较中消除弱的、低得分的比对。我们的目标是客观地选择同线性块的子集,在尊重比较基因组的复制历史的同时最大化其总得分。我们将这种方法称为基于配额的同线性块筛选,以便在一个基因组内或具有 WGD 事件的两个基因组之间适当填充同线性关系的配额。
我们已经将同线性块筛选表述为一种称为“二进制整数规划”(BIP)的优化问题,该问题可以使用现有的线性规划求解器来解决。计算机程序 QUOTA-ALIGN 通过创建一个明确的目标函数来执行此任务,该目标函数最大化给定重叠和深度约束下(对应于各自基因组中的复制历史)的同线性块兼容集。这种方法对于任何两两同线性比对都很有用,但在受多次 WGD 影响的谱系中最有用,例如植物或鱼类谱系。例如,如果基因组 B 在两个基因组分化后经历了独立的 WGD,则基因组 A 和 B 之间应该存在 1:2 的倍性关系。我们通过在蔷薇超目植物基因组中的模拟和实际示例表明,基于配额的筛选可以消除模棱两可的同线性块,并专注于特定的基因组进化事件,例如谱系的分化(在种间比较中)和最近的 WGD(在自比较中)。
QUOTA-ALIGN 算法筛选一组同线性块,只保留与两个基因组之间用户指定的倍性关系兼容的那些。这些块反过来又可以用于其他下游分析,例如在种间比较中识别真正的同源区域。QUOTA-ALIGN 的两个主要贡献是:1)将块筛选任务简化为 BIP 问题,这是新颖的;2)提供了一个从所有对所有 BLAST 到带有点图可视化的筛选同线性块的高效软件管道。Python 代码和完整文档可在 http://github.com/tanghaibao/quota-alignment 上公开获取。QUOTA-ALIGN 程序也作为 SynMap 的主要组件集成在内 http://genomevolution.com/CoGe/SynMap.pl,为非程序员提供了对数千个基因组的更简单访问。