Böckmann Ineke, Haffner Dieter
Department of Pediatric Kidney, Liver, Metabolic and Neurological Diseases, Hannover Medical School, Carl-Neuberg-Str. 1, Hannover, Germany.
Calcif Tissue Int. 2025 Apr 28;116(1):66. doi: 10.1007/s00223-025-01374-w.
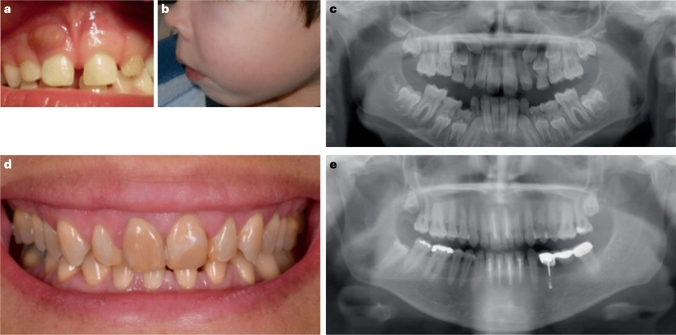
X-linked hypophosphatemia is a rare genetic disease caused by pathogenic variants in the PHEX (phosphate-regulating endopeptidase homolog X-linked) gene with X-linked dominant inheritance that causes metabolic bone disease and other severe complications. PHEX dysfunction results in increased production and secretion of the phosphaturic hormone fibroblast growth factor 23 (FGF23) from bone. The consequences of FGF23 excess are renal phosphate wasting and decreased calcitriol synthesis, leading to hypophosphatemia and subsequently rickets and osteomalacia. Children with XLH usually become symptomatic in the second year of life presenting with progressive disproportionate short stature, bone pain, frontal bossing, enlarged joints, bowed legs, and a waddling gait. Various other symptoms may develop later, including dental abscesses, peritonitis, hearing loss, pseudofractures, spinal stenosis, osteoarthritis, and enthesopathies, often leading to a diminished quality of life and ultimately disability. Here, we provide an overview of the current knowledge of the pathophysiology and treatment insights of this rare and challenging disease, including the targeting of FGF23 as a therapeutic approach that has significantly improved patient outcomes.
X连锁低磷血症是一种罕见的遗传性疾病,由PHEX(X连锁磷酸盐调节内肽酶同源物)基因的致病变异引起,具有X连锁显性遗传,可导致代谢性骨病和其他严重并发症。PHEX功能障碍导致骨中磷尿激素成纤维细胞生长因子23(FGF23)的产生和分泌增加。FGF23过量的后果是肾脏磷酸盐流失和骨化三醇合成减少,导致低磷血症,随后出现佝偻病和骨软化症。XLH患儿通常在出生后第二年出现症状,表现为进行性不成比例的身材矮小、骨痛、额部隆起、关节肿大、弓形腿和蹒跚步态。各种其他症状可能在以后出现,包括牙脓肿、腹膜炎、听力丧失、假性骨折、椎管狭窄、骨关节炎和附着点病,常常导致生活质量下降,最终导致残疾。在此,我们概述了这种罕见且具有挑战性疾病的病理生理学和治疗见解的当前知识,包括将FGF23作为一种治疗方法进行靶向治疗,这已显著改善了患者的预后。