Frankenfield Ashley M, Yang Kevin L, Mazli Wan Nur Atiqah Binti, Shih Jamison, Yu Fengchao, Lo Edwin, Nesvizhskii Alexey I, Hao Ling
Department of Chemistry, George Washington University, Washington, District of Columbia, USA.
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, USA.
Mol Cell Proteomics. 2025 Apr 30;24(6):100980. doi: 10.1016/j.mcpro.2025.100980.
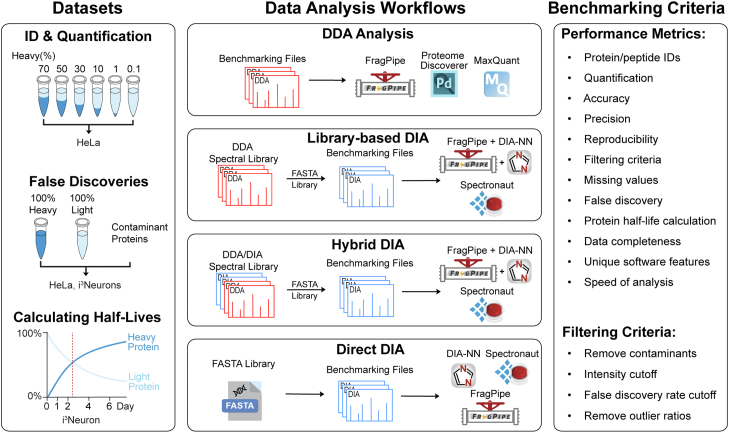
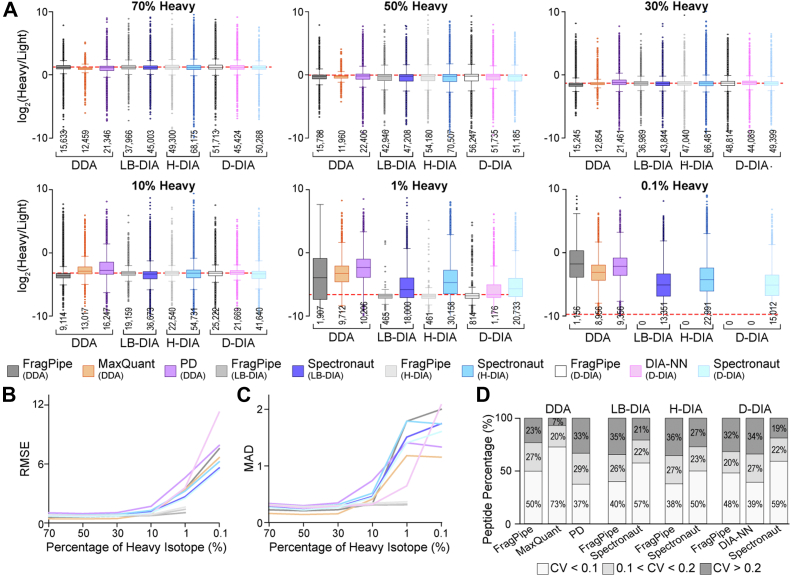
Stable isotope labeling by amino acids in cell culture (SILAC) is a powerful metabolic labeling technique with broad applications and various study designs. SILAC proteomics relies on the accurate identification and quantification of all isotopic versions of proteins and peptides during both data acquisition and analysis. However, a comprehensive comparison and evaluation of SILAC data analysis platforms is currently lacking. To address this critical gap and offer practical guidelines for SILAC proteomics data analysis, we designed a comprehensive benchmarking pipeline to evaluate various in vitro SILAC workflows and commonly used data analysis software. Ten different SILAC data analysis workflows using five software packages (MaxQuant, Proteome Discoverer, FragPipe, DIA-NN, and Spectronaut) were evaluated for static and dynamic SILAC labeling with both DDA and DIA methods. For benchmarking, we used both in-house generated and repository SILAC proteomics datasets from HeLa and neuron culture samples. We assessed 12 performance metrics for SILAC proteomics including identification, quantification, accuracy, precision, reproducibility, filtering criteria, missing values, false discovery rate, protein half-life measurement, data completeness, unique software features, and speed of data analysis. Each method/software has its strengths and weaknesses when evaluated for these performance metrics. Most software reaches a dynamic range limit of 100-fold for accurate quantification of light/heavy ratios. We do not recommend using Proteome Discoverer for SILAC DDA analysis despite its wide use in label-free proteomics. To achieve greater confidence in SILAC quantification, researchers could use more than one software packages to analyze the same dataset for cross-validation. In summary, this study offers the first systematic evaluation of various SILAC data analysis platforms, providing practical guidelines to support decision-making in SILAC proteomics study design and data analysis.
细胞培养中氨基酸稳定同位素标记(SILAC)是一种强大的代谢标记技术,具有广泛的应用和多样的研究设计。SILAC蛋白质组学依赖于在数据采集和分析过程中对蛋白质和肽的所有同位素形式进行准确鉴定和定量。然而,目前缺乏对SILAC数据分析平台的全面比较和评估。为了填补这一关键空白并为SILAC蛋白质组学数据分析提供实用指南,我们设计了一个全面的基准测试流程,以评估各种体外SILAC工作流程和常用的数据分析软件。使用五个软件包(MaxQuant、Proteome Discoverer、FragPipe、DIA-NN和Spectronaut)的十种不同SILAC数据分析工作流程,通过DDA和DIA方法对静态和动态SILAC标记进行了评估。为了进行基准测试,我们使用了来自HeLa和神经元培养样本的内部生成的和存储库中的SILAC蛋白质组学数据集。我们评估了SILAC蛋白质组学的12个性能指标,包括鉴定、定量、准确性、精密度、可重复性、过滤标准、缺失值、错误发现率、蛋白质半衰期测量、数据完整性、独特的软件功能以及数据分析速度。在评估这些性能指标时,每种方法/软件都有其优点和缺点。大多数软件在准确量化轻/重比时达到100倍的动态范围限制。尽管Proteome Discoverer在无标记蛋白质组学中广泛使用,但我们不建议将其用于SILAC DDA分析。为了在SILAC定量中获得更高的可信度,研究人员可以使用多个软件包来分析同一数据集以进行交叉验证。总之,本研究首次对各种SILAC数据分析平台进行了系统评估,提供了实用指南,以支持SILAC蛋白质组学研究设计和数据分析中的决策。