Shrestha Bibek, Kandel Grishma, Adhikari Dhiraj, Bastakoti Sudip, Mishra Abhinash
Maharajgunj Medical Campus, Tribhuvan University, Institute of Medicine, Kathmandu Nepal.
Department of Internal Medicine, Tribhuvan University Teaching Hospital, Kathmandu Nepal.
Ann Med Surg (Lond). 2025 Apr 4;87(5):3037-3042. doi: 10.1097/MS9.0000000000003261. eCollection 2025 May.
Xeroderma pigmentosum is a rare autosomal recessive genetic disorder characterized by defective nucleotide excision repair, leading to extreme sensitivity to ultraviolet radiation, skin pigmentation changes, and a heightened risk of malignancies. Neurological symptoms, such as seizures and cognitive decline, are observed in 20% of cases. While xeroderma pigmentosa is commonly associated with skin cancers, systemic malignancies like lung carcinoma are exceedingly rare.
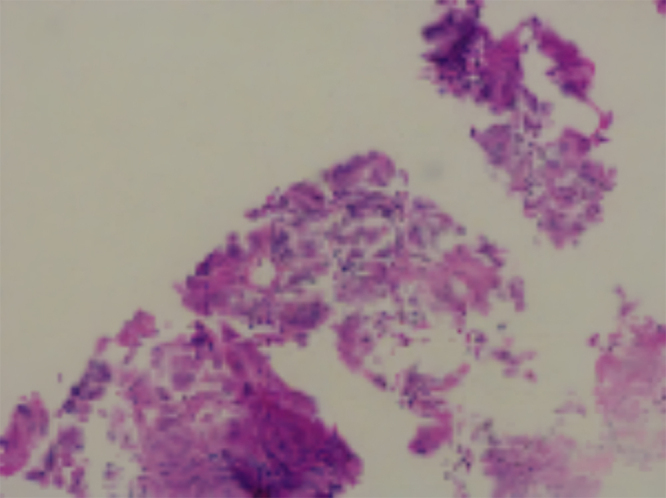
A 22-year-old male with a history of xeroderma pigmentosa presented with a six-month history of non-productive cough, hemoptysis, intermittent fever, and episodic jerky movements in his upper limb. Examination revealed hypo- and hyperpigmented macules, cachexia, cervical lymphadenopathy, and diminished air entry in the left lung. Blood tests indicated leukocytosis, elevated ESR, and abnormal electrolyte levels. Imaging confirmed a left lower lobe lung mass, and biopsy revealed spindle cell carcinoma with p53 positivity on immunohistochemistry.
Spindle cell carcinoma is a rare and aggressive subtype of non-small-cell lung cancer, comprising 0.2-0.3% of pulmonary malignancies. Its association with XP is notable, as defective DNA repair mechanisms in xeroderma pigmentosa predispose patients to malignancies driven by p53 mutations. This case emphasizes the need for vigilance for systemic malignancies in XP patients.
This is the first reported case of xeroderma pigmentosa associated with spindle cell carcinoma of the lung and focal seizures. It underscores the importance of early recognition and comprehensive surveillance for rare malignancies in XP patients, given their elevated cancer risk. The patient opted for palliative care and was symptomatically managed.
着色性干皮病是一种罕见的常染色体隐性遗传病,其特征为核苷酸切除修复缺陷,导致对紫外线辐射极度敏感、皮肤色素沉着改变以及患恶性肿瘤的风险增加。20%的病例会出现癫痫发作和认知衰退等神经症状。虽然着色性干皮病通常与皮肤癌相关,但肺癌等系统性恶性肿瘤极为罕见。
一名22岁有着色性干皮病病史的男性,出现了6个月的干咳、咯血、间歇性发热以及上肢发作性抽搐。检查发现有色素减退和色素沉着斑、恶病质、颈部淋巴结肿大以及左肺呼吸音减弱。血液检查显示白细胞增多、血沉升高以及电解质水平异常。影像学检查证实左肺下叶有肿块,活检显示为梭形细胞癌,免疫组化显示p53阳性。
梭形细胞癌是非小细胞肺癌中一种罕见且侵袭性强的亚型,占肺部恶性肿瘤的0.2 - 0.3%。它与着色性干皮病的关联值得注意,因为着色性干皮病中缺陷的DNA修复机制使患者易患由p53突变驱动的恶性肿瘤。该病例强调了对着色性干皮病患者系统性恶性肿瘤保持警惕的必要性。
这是首例报道的着色性干皮病合并肺梭形细胞癌及局灶性癫痫发作的病例。鉴于着色性干皮病患者患癌风险升高,该病例凸显了对其罕见恶性肿瘤进行早期识别和全面监测的重要性。患者选择了姑息治疗并接受了对症处理。